AIQM-PBSA:用AI势能突破蛋白质-配体结合自由能计算的精度瓶颈

AIQM-PBSA:用AI势能突破蛋白质-配体结合自由能计算的精度瓶颈

DrugIntel
发布于 2026-05-26 19:52:38
发布于 2026-05-26 19:52:38

文献来源:J. Phys. Chem. B 2026, 130, 4982–4993 DOI:10.1021/acs.jpcb.6c00306 作者:Xue-Xin Wei, Yuxinxin Chen, Yuedong Yang, Mingyuan Xu,* Pavlo O. Dral,* and Hongming Chen* 机构:广州国家实验室、中山大学、厦门大学、广州医科大学
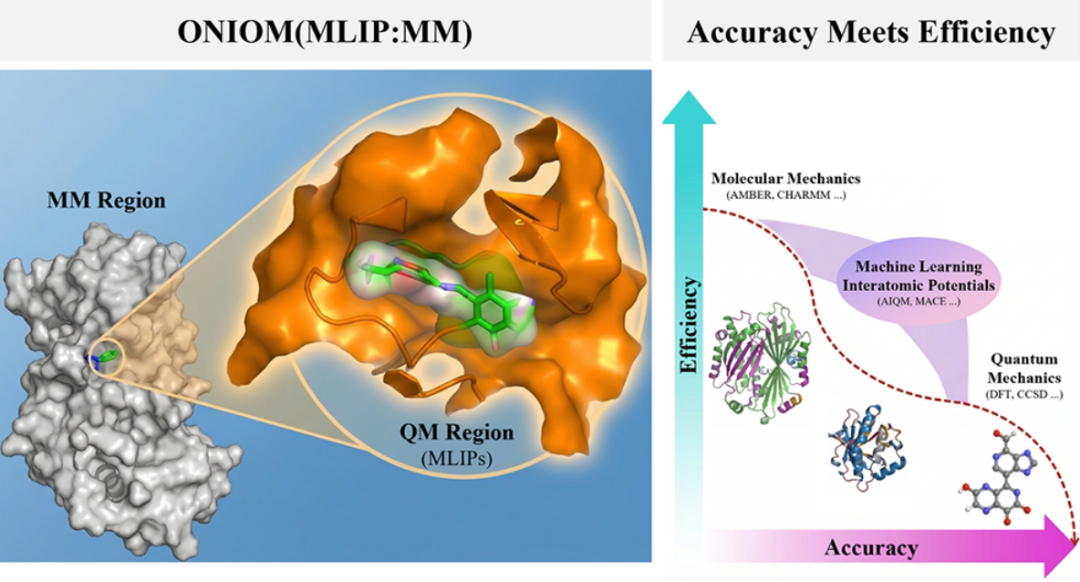
一、研究背景
1.1 结合自由能预测的核心地位
蛋白质-配体结合自由能(Binding Free Energy, BFE)是计算机辅助药物设计(CADD)的核心物理量。精准预测 BFE 意味着在实验合成前即可筛选高亲和力候选药物,大幅缩短研发周期与成本。实验测定方法(ITC、SPR 等)虽可靠,但通量低、周期长,无法满足大规模虚拟筛选的需求。
1.2 主流计算方法的格局
目前计算 BFE 的方法主要分为三个层次:
方法类别 | 代表方法 | 精度 | 计算代价 | 适用规模 |
|---|---|---|---|---|
严格自由能方法 | FEP、RBFE | 高 | 极高(数百 ns MD) | 小批量 |
端点方法 | MMPBSA、MMGBSA | 中等 | 低 | 中/大批量 |
打分函数 | Vina、Glide SP | 低 | 极低 | 超大规模 |
MMPBSA 因计算效率与精度的平衡性,成为药物设计流程中应用最广泛的 BFE 估算工具。然而,其精度天花板明显受到两类瓶颈的限制。
1.3 MMPBSA 的双重瓶颈
瓶颈一:热力学近似
MMPBSA 的统计热力学框架存在内禀近似:忽略了构象积分的精确计算,以及配体从溶液相迁移到结合口袋时丧失的平动/转动自由度(标准态自由能罚项)。这使得 BFE 的绝对值存在系统性偏差。
瓶颈二:经典力场的局限性
标准 MMPBSA 采用 AMBER、GAFF 等经典分子力场计算气相分子间相互作用能(ΔEMM)。经典力场基于点电荷模型,存在以下根本性缺陷:
- • 无法描述电子极化效应:固定点电荷无法响应局部电场变化
- • 无法刻画电荷转移:共价边界附近的电子重分布被完全忽略
- • 各向同性原子模型的失真:以卤键为典型例子,氯原子的 σ-hole 导致其电荷分布高度各向异性,经典力场将其视为均匀负电荷球,会将实际的吸引性卤键误判为排斥性相互作用
- • 对介电常数的过度依赖:为补偿极化缺失,不得不通过调节内部介电常数(εint)来经验性拟合,严重损害方法的可迁移性
二、核心方法:AIQM-PBSA 框架
2.1 方法总体架构
本文提出的 AIQM-PBSA 框架,以 MMPBSA 的热力学框架为基础,用 ONIOM(MLIP:MM)方案替换原有的纯 MM 气相能量计算,形成如下能量分解体系:
ΔGbind = ΔEAIQM/MM + ΔGpolar(PB) + ΔGnonpolar(SASA/SAV)完整的 BFE 计算方程为:
AIQM-PBSA = α₁·ΔEAIQM/MM + α₂·ΔGPB + α₃·ΔGSASA + α₄·ΔGSAV + b其中系数 α₁–α₄ 和截距 b 来自 Akkus 等前期工作中对数据集 A 的拟合,在本框架中保持固定,以保证评估的客观性。非极性溶剂化能同时考虑 SASA 和 SAV,基于尺度粒子理论(SPT),比单纯 SASA 模型更准确。
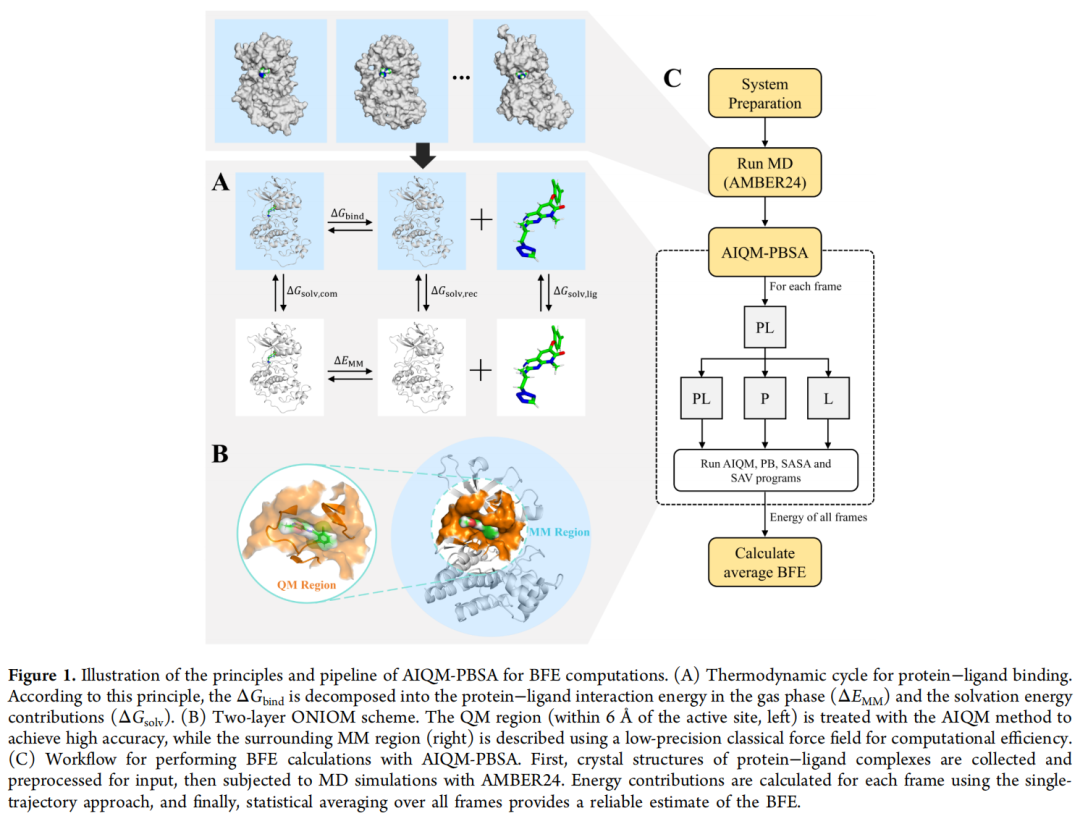
2.2 两层 ONIOM 方案
ONIOM(Our own N-layered Integrated molecular Orbital and molecular Mechanics)方案将体系划分为两个区域:
QM 区(高精度区):以配体为中心,取 6 Å 半径内所有残基,采用 AIQM3 计算。边界共价键用氢原子加帽(Hydrogen Capping)处理。
MM 区(低精度区):6 Å 以外的蛋白质环境,采用 AMBER ff99SB-ILDN 力场描述。
总能量由下式给出:
EAIQM/MM = E^QM_AIQM + E^total_MM - E^QM_MMQM-MM 边界的相互作用采用机械嵌入(Mechanical Embedding)方式处理,包括跨边界的键伸缩、角弯曲、二面角扭转势能,以及 Coulomb 和 Lennard-Jones 非键相互作用。
作者还系统比较了三种边界加帽策略(氢原子、甲基、ACE/NME 封端),发现氢原子加帽在准确性和稳定性上最优,成为默认选择。
2.3 核心势能:AIQM3
AIQM3 是 AIQM 系列的第三代模型,属于 Δ-学习 架构——在半经验量子力学(SQM)基线上叠加神经网络修正,靶向耦合簇精度:
E_AIQM3 = E_GFN2-xTB* + E_NN + E_D3(BJ)- • E_GFN2-xTB*:去除 D4 色散项的 GFN2-xTB 半经验基线,提供高效的基础能量
- • E_NN:8 个 ANI 神经网络的集成预测,从 GFN2-xTB* 水平修正至 CCSD(T)/CBS 水平
- • E_D3(BJ):显式 D3 色散修正(基于 B97-3c 泛函),改善非共价相互作用描述
两阶段 Δ-学习训练策略:
- 1. 第一阶段:在大规模 DFT(B97-3c)数据集上训练神经网络,预测从 GFN2-xTB* 到 DFT 的能量修正
- 2. 第二阶段:使用 CCSD(T)/CBS 参考数据精调,目标为从 DFT 到金标准耦合簇的残差
相比前代(AIQM1/AIQM2 仅支持 CHNO),AIQM3 新增 S、F、Cl 元素支持,完整覆盖药物分子中最常见的卤代物,使其可直接应用于蛋白质-配体体系。
三、数据集与评估体系
3.1 三套基准数据集
数据集 A(拟合集参考):5 个蛋白靶标(JNK-1、HIV-1、SARS-CoV、MERS-CoV、SARS-CoV-2),共 54 个系统。由于系数拟合时使用了该集,非严格外部测试集。
数据集 B(外部测试集):10 个蛋白靶标(Urokinase、HSP82、HSP90、Endothiapepsin、AR、β-lactamase、Pim kinase、BRD4、FXIa、PDE10A),共 24 个系统,全部来自 CASF-2016,为严格外部测试。
Schrödinger JACS 集(最严格基准):4 个激酶靶标(CDK2、JNK1、P38、TYK2),共 87 个蛋白-配体复合物。每个靶标包含多个骨架高度相似但活性跨度很大的配体,存在明显的活性悬崖(Activity Cliff),是最接近真实药物发现场景的基准。
所有配体均为中性分子,复合物仅含 H、C、N、O、F、S、Cl 七种元素。
3.2 对比方法
类别 | 方法 | 说明 |
|---|---|---|
经典 MM | MMPBSA(默认) | AMBER ff99SB-ILDN + GAFF2 |
经典 MM(优化) | PBSA_E | 对各能量项系数重拟合的优化版本 |
纯 MLIP | ANI-2x | 全系统能量,TorchANI 实现 |
纯 MLIP | MACE-OFF23(S) | 针对大体系优化的小模型 |
ONIOM(MLIP:MM) | MACE-OFF24(M) | 更高精度 MACE 模型嵌入 ONIOM |
ONIOM(MLIP:MM) | AIQM-PBSA(本文) | AIQM3 + ONIOM + PBSA |
3.3 MD 模拟参数
- • 力场:蛋白 ff99SB-ILDN,配体 GAFF2,水 TIP3P
- • 溶剂盒:cubic,10 Å buffer,Na⁺/Cl⁻ 中性化
- • 温度:310 K(NPT),压力:1 atm
- • 生产模拟:10 ns,均匀采样 1000 帧用于能量平均
- • 内部介电常数:εint = 1.0 和 2.1(外部 εout = 80)
四、主要结果与分析
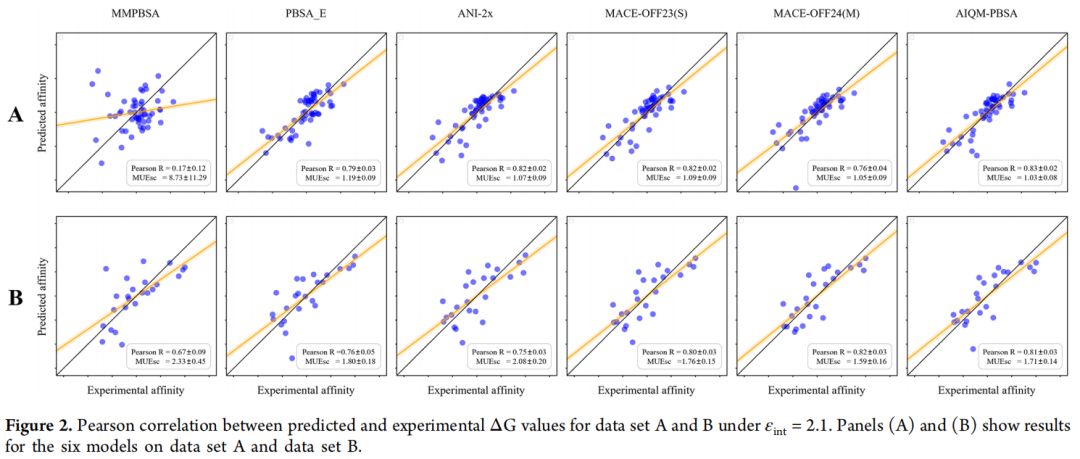
4.1 数据集 A 与 B 的总体表现
在数据集 A(εint = 1.0)上:
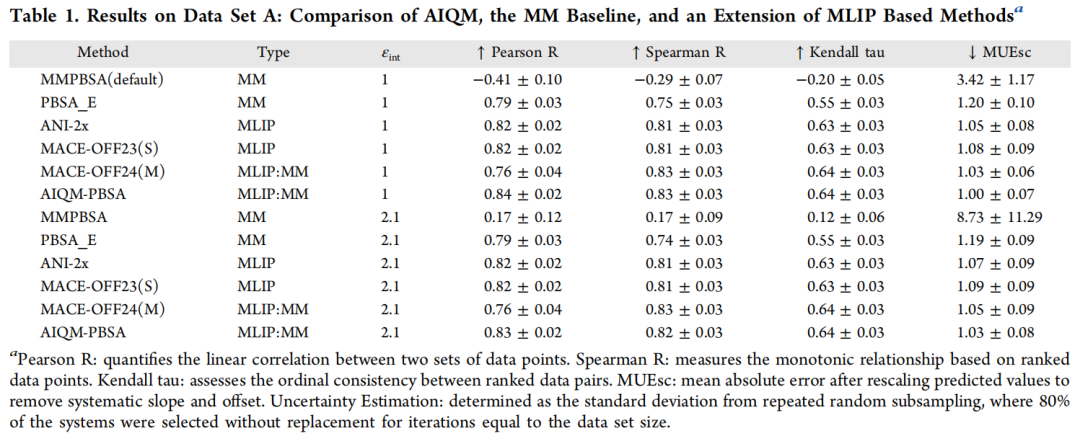
在外部测试集 B(εint = 2.1)上,AIQM-PBSA 的 Spearman R(0.85)在所有方法中最高,比 ANI-2x 高 0.10,展示了更强的排序能力——这对虚拟筛选中的化合物优先级排序至关重要。
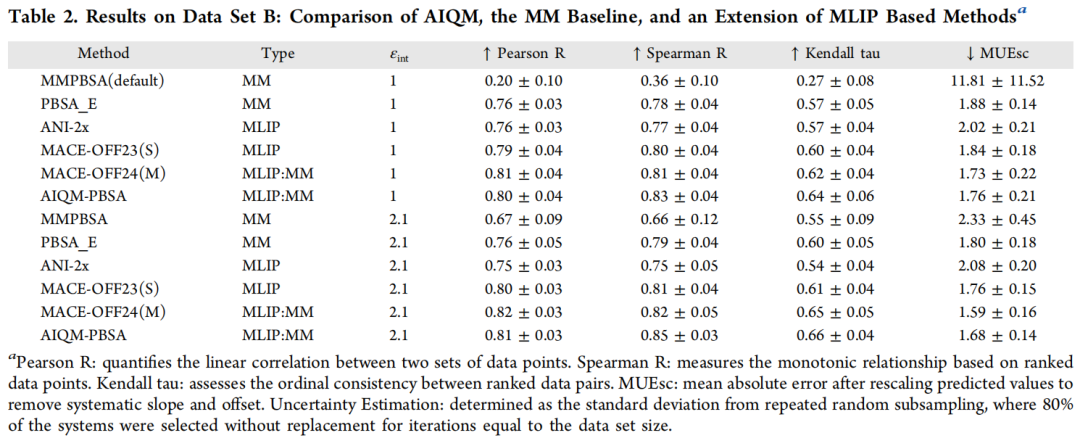
值得关注的现象:ANI-2x 在数据集 A 上接近 AIQM-PBSA(差距约 0.01),但在独立的外部测试集 B 上差距扩大到 0.06–0.10,说明 AIQM-PBSA 具有更好的泛化能力。
4.2 Schrödinger JACS 集:严格评估下的差距
该数据集是活性悬崖密集的最具挑战性基准(εint = 2.1):
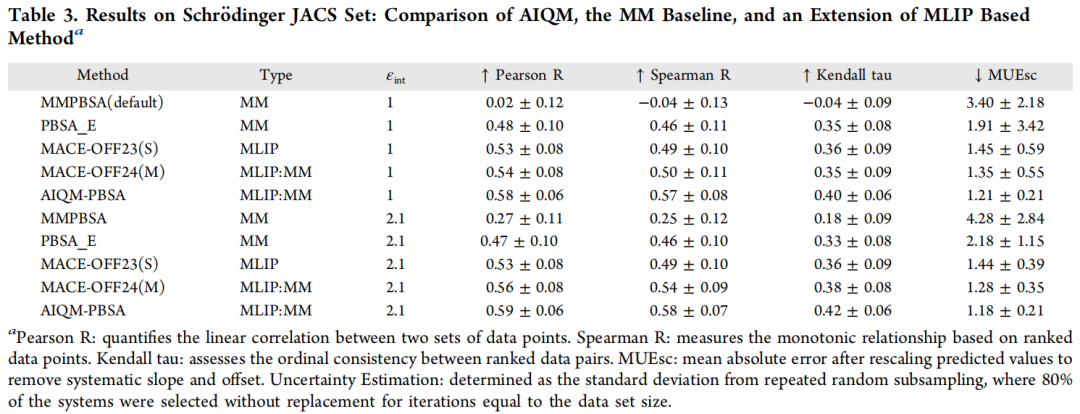
AIQM-PBSA 在 Pearson R 上比 MMPBSA 高 +0.31,比 ANI-2x 高 +0.16,差距在最严格测试场景下反而更加突出,表明其改进不是对特定数据集的过拟合,而是对物理相互作用更本质的捕捉。
从单靶标分析来看,AIQM-PBSA 在 CDK2 上优势最明显,对全部 4 个靶标的平均表现最为稳健,尽管并非在每个靶标上都是最优(这也反映了端点方法的固有局限性)。
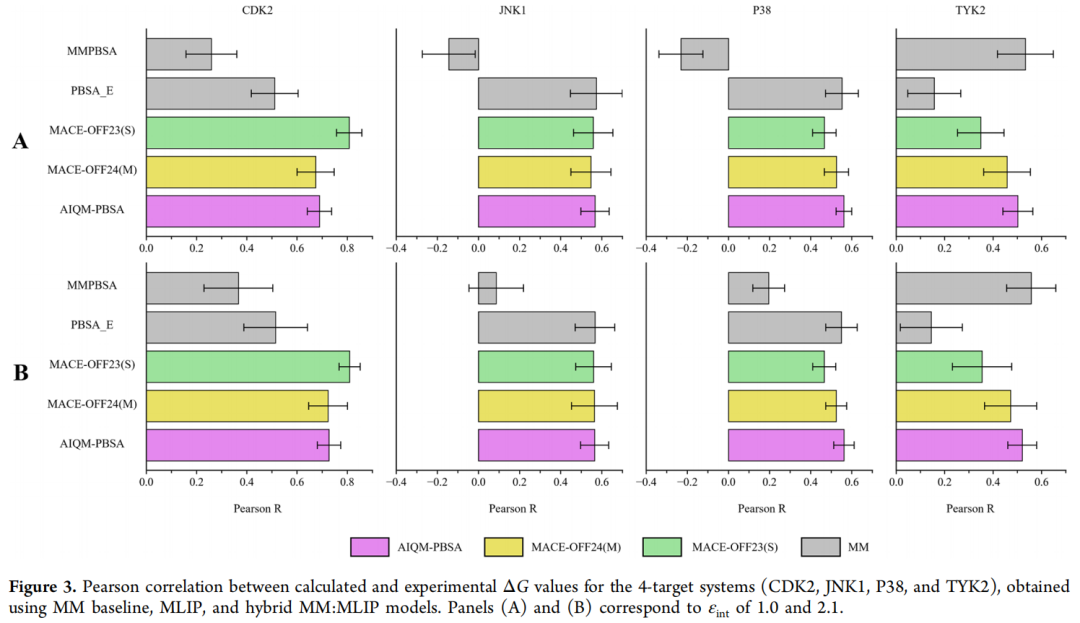
4.3 带电配体体系的泛化性
作者额外测试了 60 个带电配体复合物(来自 Zhu 等的数据集)。由于净电荷使 QM 能量尺度显著扩大,对带电体系重拟合了缩放系数(5 折交叉验证,重复 10 次)。结果显示,重拟合后的 AIQM-PBSA 精度与专门针对带电体系优化的 PBSA_E 相当,显著优于标准 MMPBSA,验证了框架对带电体系的适用性,这归功于 AIQM3 对带电物种的近 DFT 精度。
4.4 熵效应的系统评估
研究中评估了四种熵估算方法,并分析了各自的计算代价:
熵方法 | 原理 | 平均计算时间 | 整体改善效果 |
|---|---|---|---|
IE(相互作用熵) | 从气相相互作用能涨落估算熵 | 1.6 秒 | 稳定小幅提升 |
FE(公式化熵) | 基于 SASA 和可旋转键数的经验公式 | 46 秒 | 有限 |
QHA(准谐近似) | 质量加权协方差矩阵对角化 | 54.4 秒 | 体系依赖 |
NMA(简正模分析) | 局部能量极小化后 Hessian 矩阵对角化 | 约 1.8 小时 | 不稳定 |
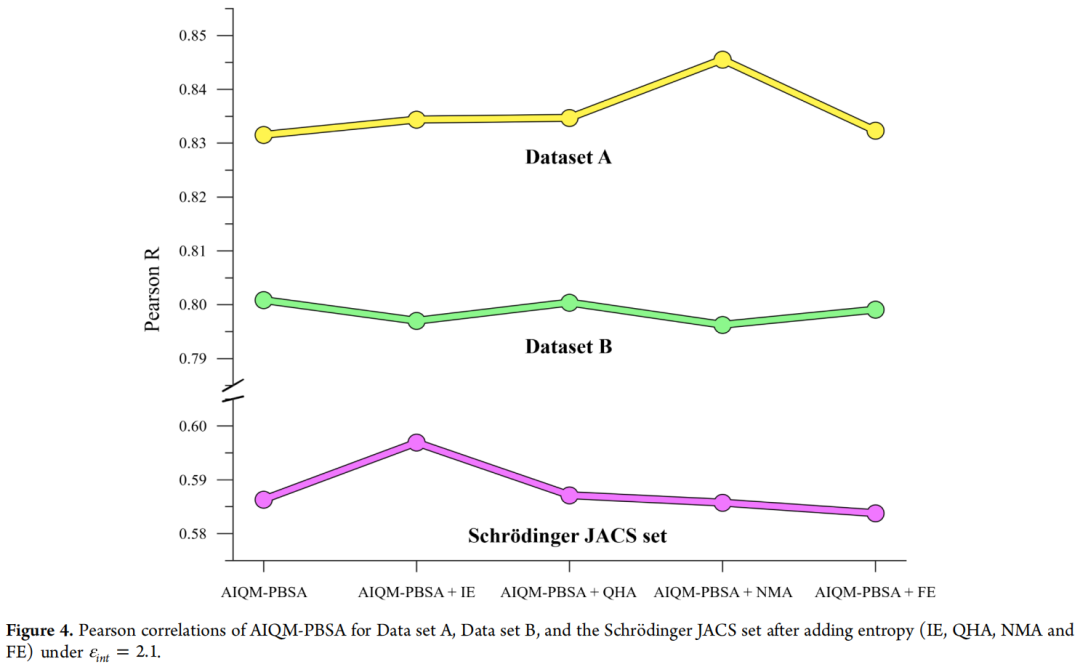
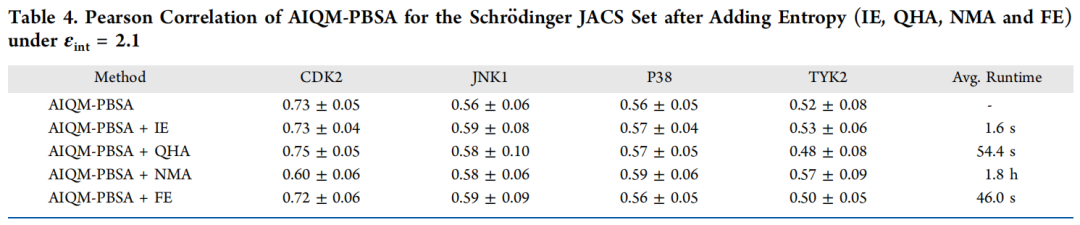
关键结论:IE 是最推荐的策略。它利用已有的气相相互作用能,不引入额外计算,在四个 JACS 靶标上均产生稳定改善(Pearson R 从 0.59 升至 0.60)。NMA 虽理论上最严格,但对 CDK2 的 Pearson R 从 0.73 骤降至 0.60,印证了 Sun 等的系统研究:熵修正的必要性高度依赖靶标口袋特征,深/柔性口袋(HIV 蛋白酶、激酶)需要熵修正,而表面活性位点(凝血酶类)加入熵反而有害。
4.5 内部介电常数的敏感性分析
作者在 εint = 1.0、2.1、3.0、5.0、10.0 五个取值下系统评估了所有方法,揭示了关键规律:
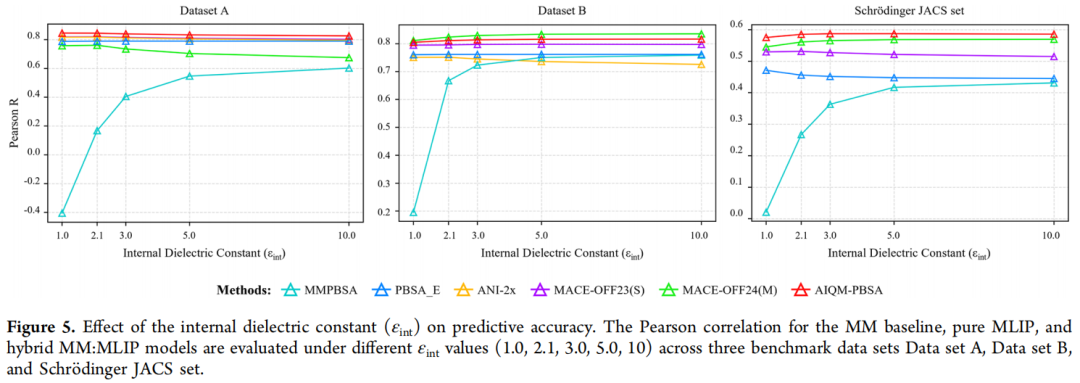
MMPBSA 极度敏感:在数据集 A 上,Pearson R 从 εint = 1.0 的 −0.40 跃升至 εint = 5.0 的 0.55,差异达 0.95。这一敏感性源于经典力场依赖固定点电荷,必须用 εint 来经验性补偿缺失的显式极化。这意味着经典 MMPBSA 的"最优"结果本质上依赖参数调优,不具有普适性。
MLIP 方法高度稳定:ANI-2x、MACE 系列、AIQM-PBSA 对 εint 变化几乎不敏感(各数据集变化 <0.02)。原因在于 MLIP 的 QM 训练势能已经内禀地编码了极化和电荷转移效应,无需依赖 εint 进行经验补偿。
这一结果具有深远意义:MLIP 框架消除了 MMPBSA 的一个重要调参自由度,使方法更加物理透明、结果更具可重复性。
五、物理机制:为什么 MLIP 能更好?
5.1 典型案例:卤键与 σ-hole 效应
以 TYK2 靶标的复合物 4GIH_ejm31 为例进行深度剖析:
该复合物的结合关键在于 C−Cl···O 卤键。其物理本质是 σ-hole 效应:
- • 碳的吸电子性使氯的电子云沿 C−Cl 轴方向被极化
- • 在 C−Cl 键轴方向出现带正电的"σ 空穴"(电子密度降低区)
- • σ 空穴与骨架羰基氧(电负性)形成高度方向性的吸引作用
- • 卤键键长约 3.2 Å,远小于范德华半径之和(约 3.3 Å)
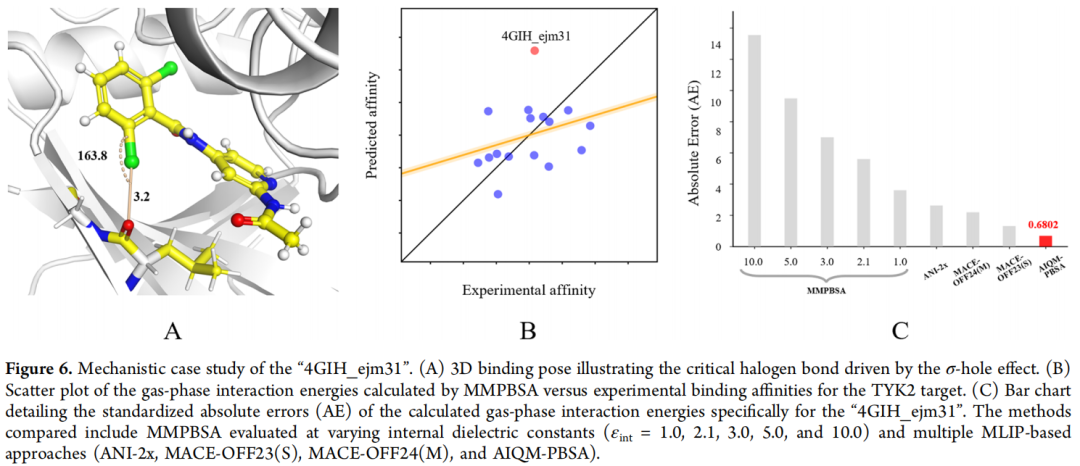
经典力场的失败:
整个氯原子被视为带均匀负部分电荷的各向同性球。当带负电的"氯"接近带负电的"羰基氧"时,力场计算出强烈的静电排斥,严重低估结合能,将该化合物预测为弱结合剂——与实验结果完全相反,使其成为 MMPBSA 最大的异常值。
诡异的介电常数依赖:
对于普通体系,较大的 εint(如 5.0)通常更优(通过稀释静电项来降低力场噪声)。但对 4GIH_ejm31,恰恰是较小的 εint(1.0)反而给出"较好"结果——原因是小 εint 放大了负静电项,在这个特殊案例中阴差阳错地减小了绝对误差。这种案例特异的"最优"参数,暴露了经典方法的根本不稳健性。
AIQM-PBSA 的正确描述:
AIQM3 从第一性原理出发,明确描述卤键的各向异性电荷分布,σ-hole 的方向性静电吸引被正确量化,误差显著低于所有经典方法,且不随 εint 变化。
六、计算效率分析
计算模块 | 硬件 | 单帧耗时 |
|---|---|---|
AMBER 经典力场 | CPU(单核) | 秒级 |
ANI-2x(全系统) | GPU(A100) | 秒级 |
MACE-OFF23(S)(全系统) | GPU(A100) | 秒级 |
AIQM3(6 Å 口袋,~570 原子) | CPU(单核) | ~83 秒 |
在典型的 64 核 + 单 GPU 生产环境中,用 MLIP 替换经典 MM 后的额外时间开销仅为总流程的 2%–10%,完全在可接受范围内。相比传统全系统 DFT 计算(原子数相同时需数小时),效率提升超过 3 个数量级。
七、局限性与展望
7.1 当前局限
- 1. 长程相互作用处理不足:ONIOM 的机械嵌入将 QM-MM 界面的长程极化效应简化为点电荷相互作用,静电嵌入方案有望进一步提升精度但代价更高
- 2. 隐式溶剂化模型的近似:PBSA 模型对溶剂结构的描述仍有局限,特别是对结合口袋中明确水分子的处理不够精确
- 3. 熵估算方法有限:现有四种熵方法均存在精度或效率的权衡,熵的精确计算仍是 BFE 方法的根本难题
- 4. 系数拟合的数据依赖性:AIQM-PBSA 的线性系数(α₁–α₄)拟合于特定数据集,跨靶标类别的泛化仍需验证
7.2 未来方向
- • 集成更先进的 MLIP:框架设计允许无缝替换 AIQM3 为更新一代势能(如 AIQM4 或专用生物分子 MLIP),精度上限持续提升
- • 静电嵌入替代机械嵌入:在 QM-MM 边界引入极化效应,进一步减小边界误差
- • 显式溶剂分子与自由能微扰结合:将端点方法的高效性与 FEP 的严格性结合,在关键步骤引入更高精度
- • 机器学习熵估算:基于大规模 MD 数据训练熵预测模型,取代现有的经验或简谐近似方法
小结
AIQM-PBSA 代表了一类重要的方法论范式:不颠覆已有流程,而是通过替换核心计算模块(从经典力场到量子级 MLIP)实现精度的阶跃式提升。这种"即插即用"的设计哲学降低了采用门槛,使工业界用户可以在现有 MMPBSA 流程中以最小代价切换至更高精度方案。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-23,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号