数据才是第一生产力:人工智能驱动小分子药物发现的困境与出路

数据才是第一生产力:人工智能驱动小分子药物发现的困境与出路

DrugIntel
发布于 2026-05-29 12:59:40
发布于 2026-05-29 12:59:40

文献推荐 | Nature Computational Science, 2024 Durant G, Boyles F, Birchall K & Deane CM. "The future of machine learning for small-molecule drug discovery will be driven by data." Nat Comput Sci (2024). https://doi.org/10.1038/s43588-024-00699-0 作者单位:英国牛津大学统计系、LifeArc
导读:一篇「泼冷水」的重要综述
近年来,机器学习(ML)在小分子药物发现领域的应用引发了广泛关注与期待。然而,来自牛津大学 Charlotte Deane 课题组的这篇 Perspective,以罕见的冷静视角审视了这一领域的真实进展——算法在进步,但结果并没有跟上。
作者的核心论断直截了当:在小分子药物发现中,制约 ML 方法性能提升的主要瓶颈,并非算法架构的先进程度,而是训练与验证所用数据的质量与数量。这一判断,对当前大量追逐模型新颖性的研究范式,是一次重要的校正。
一、背景:药物发现的困境与 ML 的期望
1.1 小分子药物的重要性与开发挑战
小分子化合物占全球已批准药物的 90%,是现代医学的核心支柱。它们分子量低,能穿透细胞膜,直接调控蛋白质或 RNA 靶点的活性。近年来,其应用范式还在不断拓展,包括蛋白质降解靶向嵌合体(PROTACs)和分子胶水等新兴模式。
然而,小分子药物的研发面临严峻挑战。自 1950 年至 2012 年,每花费 10 亿美元获批的新药数量,每 9 年减少一半,这一趋势被称为「Eroom 定律」(Moore 定律的逆转)。一款新药从研发到上市,往往历时数年乃至十年,耗资数亿至数十亿美元。
1.2 ML 被寄予厚望的理由
ML 在以下邻近领域的成功,为药物发现带来了想象空间:
- • 计算机视觉:扩散模型、对比学习等技术催生了 DALL-E、Stable Diffusion 等突破
- • 自然语言处理:GPT-4、LLaMA 2 等大语言模型涌现出接近人类水平的能力
- • 蛋白质结构预测:AlphaFold 2 在 CASP14 竞赛中比第二名精度高出 2.6 倍,被誉为「结构生物学的 AlphaFold 时刻」
上述成功的共同特征是:数据丰富 + 精良架构,两者缺一不可。
在小分子药物发现中,ML 已渗透多个子领域:
任务 | 描述 |
|---|---|
从头分子设计(De novo design) | 生成具有目标性质的新颖分子 |
逆合成预测(Retrosynthesis) | 规划高效合成路线 |
分子对接(Docking) | 预测配体与靶点结合的三维构象 |
性质预测(Property prediction) | 估计 ADMET、活性等关键分子性质 |
二、核心发现:算法升级并未带来性能跃升
2.1 基准测试揭示的残酷现实
作者收集了三个经典基准测试,将历年文献的性能指标与发表时间进行相关分析(Pearson's R),结果如下:
基准测试 | 任务 | 指标 | 时间-性能相关性(ρ) | 结论 |
|---|---|---|---|---|
CASF-2016 | 蛋白质-配体结合亲和力预测 | Pearson's R | 0.03 | 近乎零相关,无进展 |
HIV MoleculeNet | HIV 蛋白酶活性分类(QSAR) | AUROC | 0.09 | 近乎零相关,无进展 |
USPTO-50k | 单步逆合成预测 | Top-1 准确率 | 0.87 | 有增长,但仍无质变 |
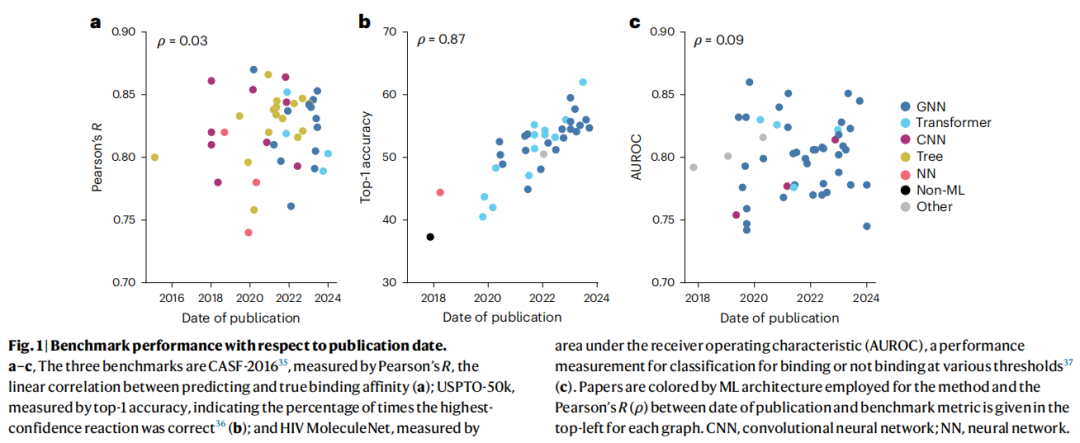
值得注意的是,即便是表现出最明显提升趋势的 USPTO-50k,也仅是渐进式的,远未达到 AlphaFold 2 那种数量级的跃升。
2.2 先进架构为何「水土不服」?
图神经网络(GNN) 理论上非常适合小分子:它可以直接编码分子图结构,无需手工特征化。GNN 在材料发现(GNoME)、新型抗生素发现等领域已有成功案例。然而,在小分子药物发现的全面评测中,GNN 相比传统方法并无系统性优势。
等变图神经网络(Equivariant GNN) 将三维对称性(旋转、平移不变性)直接嵌入网络结构,省去了数据增强的需求。但研究表明,这一架构进步同样未能在精度或泛化性上带来显著提升,其主要价值在于提供了更可解释的预测归因。
扩散模型(Diffusion models) 在图像生成领域大放异彩(DALL-E、Midjourney)。当其被应用于分子对接(DiffDock)和条件式从头分子生成时,尽管相比旧方法精度有所提高,但两项独立研究(PoseBusters、PoseCheck)均揭示:这些模型生成的分子构象在物理化学层面并不合理,与蛋白质的相互作用也存在明显缺陷。
作者的结论是:未来令人期待的新方法(如一致性模型 Consistency models、流匹配 Flow matching),如果仅以「开箱即用」的方式应用,同样不太可能带来质的飞跃。真正的进步,需要架构创新、工程决策与数据质量三者的协同。
三、数据量问题:根本性的规模鸿沟
3.1 小分子数据的先天不足
ML 的本质是从数据中学习规律,数据量越大,模型越强——这一定律在小分子领域面临严峻的现实壁垒:
- • 实验数据生成成本高昂、劳动密集,难以实现像互联网数据那样的被动式大规模积累
- • 制药公司将大量数据作为核心知识产权严格保密,形成数据孤岛
- • 公开数据集规模有限,通常在数万至数十万量级
数量级对比:
小分子 ML 数据集: 数万 ~ 数十万
--------------------------------------------
蛋白质序列数据库(UniRef): 数亿
图像生成训练数据(DALL-E 2):数亿
大语言模型训练数据(LLaMA 2):数万亿 token3.2 结构数据的特殊困境
对接算法、结构生成模型和打分函数,都依赖蛋白质-配体复合物的三维实验结构。
PDBBind 是最重要的公开结构数据集,包含所有能找到结合亲和力标注的 PDB 结构,2020版本仅收录 19,443 个蛋白质-配体复合物。这一规模构成了相关模型的性能天花板——已有研究表明,扩大数据量和覆盖度可以显著提升模型性能。
高通量结构测定(如高通量 X 射线晶体学)目前主要用于片段筛选,数据多样性有限。因此,短期内大幅扩充结构数据量或多样性的可能性不大。
当前的数据扩充策略包括:
- • 交叉对接增强:使用与晶体配体 RMSD 较低的交叉对接构象扩充训练数据
- • 合成配体生成:将蛋白质链的氨基酸残基当作独立配体处理
- • 合成口袋增强:基于 PDBBind 中相互作用频率统计生成新的结合口袋
- • 自蒸馏(Self-distillation):利用置信度校准良好的算法(参考 AlphaFold 2 的思路)生成伪标签
- • 基于物理方法的预训练:利用分子力学、量子化学计算结果预训练模型
3.3 负面数据的严重缺失
在小分子研究中,负面数据(无活性化合物、失败反应)几乎不公开,原因是多方面的:
- 1. 发表偏倚:期刊倾向发表阳性结果
- 2. 知识产权保护:失败实验数据同样可能泄露研究方向
- 3. 数据整理成本高:负面数据往往分散、不规范,整理难度大
这导致训练数据中正负样本严重失衡,例如在产率预测任务中,模型无法学习到失败反应的规律,预测可靠性大打折扣。
潜在解决路径:
- • 学术发表规范的改革(强制要求附上负面数据)
- • 合成负样本生成
- • 从博士论文、实验室记录中通过 ML 方法挖掘暗数据
- • ChemDataExtractor:从文献中自动提取化学信息
- • DECIMER.ai:光学化学结构识别(图像 → 结构式)
- • Nougat:将科学文献图像转换为标记语言
3.4 新兴数据来源
众包药物发现:COVID Moonshot 项目是一个标杆案例。该项目针对新冠病毒 Mpro 蛋白酶,以完全开放的协作方式推进药物发现,最终产出:
- • 470 个晶体结构
- • 2,000+ 化合物 的 IC₅₀ 活性数据
- • 3,000+ 化合物 的合成数据
所有数据完全公开,极大丰富了公开数据资源。
联邦学习(Federated Learning):在不转移原始数据的前提下,允许各机构共享模型知识,从而在保护知识产权的同时实现数据协作。两个典型项目:
- • MELLODDY 项目(欧盟多家制药公司参与):模型适用域平均扩展 10%
- • Effiris(Lhasa Limited 开发):适用域扩展高达 83%
四、数据质量问题:噪声、偏差与假阳性
4.1 数据偏差:模型学到了「幻象」
ML 模型会忠实地学习训练数据中的所有规律——包括那些并不反映真实物理化学规律的「幻象」模式,即数据偏差(Data bias)。
人为偏差(Anthropogenic bias):数据并非随机采样,而是由研究者主动选择,往往偏向熟悉的化学空间和合成路线。例如,药物化学家对某些合成路径的偏好,更多源于习惯而非效率。
归纳偏差(Inductive bias):最典型的例子是基于 ML 的虚拟筛选工具。多项研究表明,这类工具往往只依赖配体层面的特征(本质上是配体-配体相似性)而非真正学习蛋白质-配体相互作用的规律。
研究者提出了不对称验证嵌入(Asymmetric validation embedding)来缓解这一问题:通过构造更严苛的训练-验证集划分,惩罚「背配体」的模型。结果发现,许多被广泛引用的方法,其真实准确率远低于论文中报告的数值。
4.2 数据噪声:标签的不确定性被低估
小分子数据集中存在大量噪声,主要来源包括:
实验条件不一致:
最典型的是将来自不同实验室、不同测定条件的 IC₅₀ 数据混合,训练单一 QSAR 模型。问题在于,不同测定条件使这些数据并不严格可比:
- • 同一靶点、同一化合物,来自不同实验室的 IC₅₀ 值之间,R² 仅为 0.53
- • 即便是相对更一致的 Kᵢ 值,R² 也仅为 0.81
这意味着标签本身存在相当大的不确定性。研究估计,CASF-2016 上 ML 评分函数的理论精度上限仅为 Pearson's R 0.76,而这一上限并非源于模型缺陷,而是数据噪声的固有极限。
假阳性:泛筛干扰化合物(PAINs):
PAINs 是一类通过干扰检测机制(氧化还原循环、聚集等)而非真正与靶点结合,从而产生假阳性信号的化合物。
令人震惊的是,作者指出:广泛使用的 HIV MoleculeNet 数据集中,404 个「真实活性」化合物里,70% 命中了 Brenk 或 NIH 的 PAINs 子结构过滤规则。这意味着该基准测试中相当比例的「真实信号」可能是实验噪声。
另一个案例:在常用的元学习基准中,仅凭一个化合物命中多少靶点(PAINs 的典型特征)就能高度预测其「活性」,说明数据集本身被噪声严重污染。
五、验证体系的问题:自我感觉良好的幻觉
5.1 现有验证范式的系统性缺陷
当前小分子 ML 方法的验证,通常遵循固定模式:
- 1. 提出新架构
- 2. 在流行训练集上训练
- 3. 在流行基准测试集上评估
- 4. 报告相对于旧方法的(通常微小的)提升
这一范式存在根本性问题:测试数据往往与训练数据同质,而模型在真实药物发现中面对的是分布外(OOD)数据。
两个典型案例:
案例一:分子对接的「重对接」问题
ML 对接方法通常以「重对接(Redocking)」为评测标准:给定蛋白质-配体复合物的准确晶体结构,预测配体的结合构象。然而,能进行重对接的前提是晶体结构已知——如果结构已知,还需要对接做什么?
当研究者在更真实的场景下评测(如对接入预测蛋白结构或不准确的构象),模型性能大幅下滑。重对接给出的是过于乐观的精度估计。
案例二:逆合成的单步 vs 多步脱节
USPTO-50k 基准评测单步反应预测准确率,但实际使用时,逆合成工具需要配合蒙特卡洛树搜索(MCTS)等路径搜索算法,找到完整的多步合成路径。
一项最新研究发现:单步方法在 USPTO-50k 上的准确率,与其在完整多步路径搜索中的成功率之间存在明显脱节,且该基准的规模也被认为对于评测在更大更多样数据集上训练的模型而言太小。
5.2 改进方向
基线测试与消融实验:
作者展示了一个令人深思的例子:简单的基于决策树的评分函数,仅使用配体或蛋白质层面的特征(即只能学习数据集偏差),在多个基准上与复杂的 GNN 方法性能相当。这说明,相当一部分「模型进步」其实来自于学习数据集偏差,而非真正理解物理化学规律。
消融实验(Ablation study)应更系统地推广,明确区分架构改进、数据改进、训练策略改进各自的贡献。AlphaFold 2 的成功很大程度上归功于其详尽的消融实验报告,证明了每个创新组件的独立价值。
更严格的基准体系:
机制 | 代表项目 | 特点 |
|---|---|---|
在线排行榜 | Therapeutic Data Commons (TDC) | 集中跟踪发表进展,提供细粒度任务分析 |
盲测竞赛 | Drug Design Data Resource (D3R) | 验证集保密,避免过拟合基准 |
盲测竞赛 | CACHE(命中筛选计算方法评估) | 公开结果,支持方法横向比较 |
六、与 AlphaFold 2 的对比:为何差距如此悬殊?
AlphaFold 2 的成功并非源于单一的关键创新,而是多个精心设计的组件协同作用的结果:
- • 不变点注意力(Invariant Point Attention):专门针对蛋白质结构问题设计的空间注意力机制
- • 迭代精化(Recycling):多轮迭代优化结构预测
- • 利用序列数据补偿结构数据的不足:蛋白质序列数据库(UniRef、MGnify)提供了数亿条序列,远超实验解析结构数量,进化信息被有效利用
而在小分子领域,类似的「杠杆数据」尚不存在:没有一个信息丰富、规模庞大的代理数据集,可以让模型通过间接学习来补偿直接实验数据的稀缺。
这是小分子 ML 与蛋白质结构预测最根本的差异所在。
七、总结与展望
核心论断
方面 | 现状 | 作者建议 |
|---|---|---|
算法 | 新架构不断涌现,但性能提升有限 | 与其追逐新架构,不如深入分析现有方法的瓶颈 |
数据量 | 公开数据规模远不足以支撑深度学习 | 推进联邦学习、众包、暗数据挖掘 |
数据质量 | 噪声高、偏差多、负样本缺失 | 建立更严格的数据清洗流程,纳入不确定性建模 |
验证体系 | 基准测试与实际应用脱节 | 推广消融测试、盲测竞赛、更真实的测试集 |
对研究者的启示
- 1. 在发表新方法前,应认真思考:性能提升来自架构创新,还是更多/更好的数据?还是仅仅学习了更多的数据集偏差?
- 2. 负面结果同样有价值:记录并发布失败实验,对推进领域共识至关重要。
- 3. 计算研究者与实验研究者需要更紧密的合作:许多数据质量问题(如 PAINs、实验条件差异)需要领域专知才能识别和处理。
- 4. 将「是否在分布外数据上泛化」作为核心评测标准:在真实药物发现场景中,模型几乎总是面对分布外数据。
写在最后
这篇文章的价值不在于提出了革命性的新方法,而在于以严谨的数据分析揭示了一个常被忽视的事实:在小分子药物发现领域,我们对机器学习的期望,远远超前于支撑这些期望所需的数据基础设施。
算法的进步是必要的,但不是充分的。如果领域内的资源继续主要流向追逐更新颖的架构,而忽视数据生态系统的建设,那么「AlphaFold 时刻」在小分子药物发现中或许还要等待很久。
数据驱动,才是正道。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-29,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号