AAAI | AI 扩散分子生成走向动态口袋:Apo2Mol 的方法逻辑、实验结果与局限

AAAI | AI 扩散分子生成走向动态口袋:Apo2Mol 的方法逻辑、实验结果与局限

DrugIntel
发布于 2026-07-13 16:22:00
发布于 2026-07-13 16:22:00

文献来源
论文题目: Apo2Mol: 3D Molecule Generation via Dynamic Pocket-Aware Diffusion Models 作者: Xinzhe Zheng, Shiyu Jiang, Gustavo Seabra, Chenglong Li, Yanjun Li 单位: University of Florida 会议: AAAI-26 代码: https://github.com/AIDD-LiLab/Apo2Mol 核心贡献一句话: Apo2Mol 将 apo 蛋白口袋到 holo 蛋白口袋的构象变化纳入 3D 分子扩散生成过程,在输入未结合态蛋白结构时,同时生成候选小分子及其诱导后的结合口袋构象。
导读
多数基于结构的分子生成模型默认蛋白结合口袋是刚性的,通常在 holo 结构上生成配体。但真实药物发现中,很多靶点只有 apo 结构,配体结合又常伴随侧链、局部骨架甚至口袋形状变化。Apo2Mol 试图处理这一错配:它构建了 24,601 个实验解析的 apo-holo 结构对,并设计一个全原子、层级图、SE(3) 等变扩散模型,从 apo 口袋出发联合生成 ligand 与 refined holo pocket。它的价值不只在于指标提升,更在于把小分子生成从静态口袋条件生成推进到口袋构象共同建模。本文将详细拆解 Apo2Mol 的问题定义、数据构建、扩散过程、网络架构、损失函数、实验结果与真实应用局限。
为什么这篇论文值得关注?
基于结构的药物设计中的一个长期矛盾是:模型喜欢确定的结构,蛋白却是动态的。
在传统 SBDD 流程中,我们通常拿一个蛋白结构作为受体,围绕结合口袋进行对接、虚拟筛选或分子生成。深度生成模型延续了这个习惯:给定一个口袋,生成一个能放进去、打分不错、性质合理的小分子。问题在于,这个口袋往往被当成静态对象。
但真实分子识别不是这样。配体结合可能导致侧链旋转、局部 loop 移动、口袋开合、疏水区域重排、氢键网络重新组织。很多靶点在早期项目中只有 apo 结构,没有配体结合后的 holo 结构。如果模型只能围绕某一个固定口袋生成分子,它可能错过那些需要诱导契合才能形成稳定结合的配体。
Apo2Mol 的切入点非常明确:不要只生成 ligand,也要同时预测 ligand 结合后蛋白口袋会怎么变。论文指出,现有多数 SBDD 生成方法仍主要基于静态 holo 结构训练或使用固定口袋进行生成,而 DynamicFlow 这类方法虽然引入了蛋白动态,但依赖 MD 轨迹;MD 成本高,并可能受力场、采样时间尺度和模拟偏差影响。Apo2Mol 选择另一条路线:直接从实验解析的 apo-holo 结构对中学习结合诱导的结构变化。
这使它成为一篇值得关注的 AI 制药方法论文:它不是单纯换一个网络模块,而是在重新定义 3D 分子生成中的条件对象。输入不再只是一个固定口袋,而是一个可能被配体诱导改变的 apo 状态。
研究背景
蛋白柔性难,不是因为大家不知道蛋白会动,而是因为它很难被高效、稳定地放进生成模型。
可以把问题拆成三个层面。
第一,结构状态不唯一。 一个蛋白可能有多个可结合构象。apo 结构、holo 结构、不同配体诱导的结构、晶体堆积影响下的结构,可能都不完全一样。模型如果只学习某个 holo 构象,会把一个动态分布压缩成单点条件。
第二,配体与蛋白变化是耦合的。 口袋不是先独立变成 holo,再放进配体。真实过程中,配体形状、电荷、氢键供受体、疏水面和体积都会影响口袋如何调整。生成 ligand 与生成 pocket conformation 应该放在同一个条件分布里,而不是分两步独立处理。
第三,直接预测全原子蛋白坐标容易破坏结构。 如果让模型像生成小分子一样直接生成蛋白口袋所有原子坐标,很容易出现键长、键角、残基几何、侧链构象不合理的问题。蛋白结构有更强的层级约束:主链框架、残基局部坐标、侧链 χ 角、残基间几何关系都需要被保留。
Apo2Mol 对这些难点的处理是:配体仍以原子类型和 3D 坐标生成;蛋白口袋不直接逐原子生成,而是在残基层面预测从 apo 到 holo 的平移、旋转和侧链 χ 角更新,从而尽量保留蛋白结构完整性。论文将学习目标定义为给定 apo pocket,建模 holo pocket 与 ligand 的联合条件分布,即 p(PH, M | PA)。
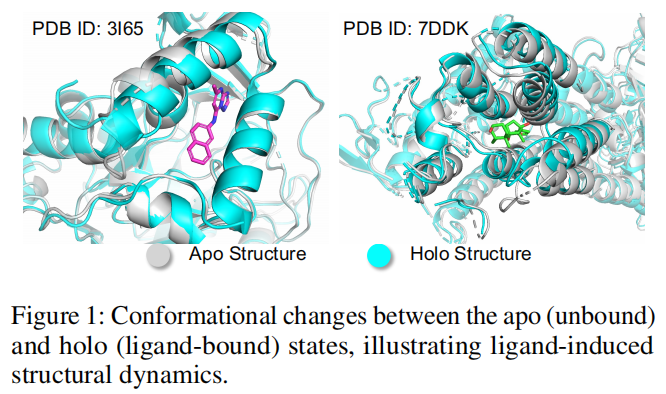
以往方法
1D/2D 分子生成:化学表示简单,但空间约束弱
早期靶点条件生成方法多基于 SMILES 或 2D molecular graph。它们可以学习化学语法、分子片段组合和性质约束,但对 3D 结合姿势、空间互补、立体冲突和口袋几何的表达有限。对于 SBDD 来说,只生成一个分子图还不够,还要知道它是否能以合理构象进入口袋并形成相互作用。
3D 分子生成:能进入空间,但常默认口袋静止
Pocket2Mol、TargetDiff、DecompDiff、IPDiff 等方法把分子生成推进到 3D 空间。它们通常在蛋白口袋条件下生成原子坐标和原子类型,再通过后处理形成键。扩散模型进一步让生成过程变成从噪声逐步去噪到合理 ligand pose 的过程。论文相关工作部分也梳理了这些方法,包括 TargetDiff 的 3D 等变扩散、DecompDiff 的 scaffold-arm 分解,以及 IPDiff 的蛋白-配体相互作用先验。
这些方法的主要限制是:口袋通常被当成静态条件。模型学习的是 ligand 如何适应口袋,而不是 ligand 与 pocket 如何共同调整。
引入动态的方法:MD 有物理细节,但成本与偏差都高
DynamicFlow 这类方法开始尝试从 apo 结构生成 holo pocket 与 ligand,但它依赖 MD 轨迹。MD 的优势是提供连续动态过程,缺点也很明显:计算成本高,轨迹能否覆盖真实结合相关构象取决于力场、采样长度和初始条件。对于大规模生成模型训练来说,MD 数据既昂贵,也可能带来模拟体系自身的偏差。
Apo2Mol 的替代思路是直接使用 PDB 中实验解析的 apo-holo 结构对。它没有真实结合路径,但有实验端点。换句话说,它不学习完整动力学轨迹,而是学习从 apo 端点到 holo 端点的结构变化分布。
这篇论文的核心思想
Apo2Mol 的核心思想可以概括为:
把 apo 到 holo 的口袋变化作为扩散生成过程的一部分,让模型在生成小分子的同时,预测这个小分子可能诱导出的结合口袋构象。
这句话包含三个关键点。
第一,输入是 apo pocket,而不是 holo pocket。对实际药物发现来说,这更接近很多早期靶点的情况。
第二,输出不是单独的小分子,而是 ligand + refined pocket。模型不仅要生成一个分子,还要给出一个从 apo 调整后的 holo-like pocket。
第三,训练数据不是 MD 模拟轨迹,而是实验解析的 apo-holo pair。论文基于 PLINDER 和 PDB 筛选得到 24,601 个 apo-holo-ligand triplets,要求 apo-holo 结构具有 100% 序列一致性、结构分辨率不高于 2.5 Å,并排除离子、辅因子、artifact、片段等不适合作为药物样小分子的配体。数据集按时间划分为 23,052 个训练样本、1,071 个验证样本和 478 个测试样本。
这套设计使 Apo2Mol 不只是 ligand generator,而是一个 dynamic pocket-aware generator。
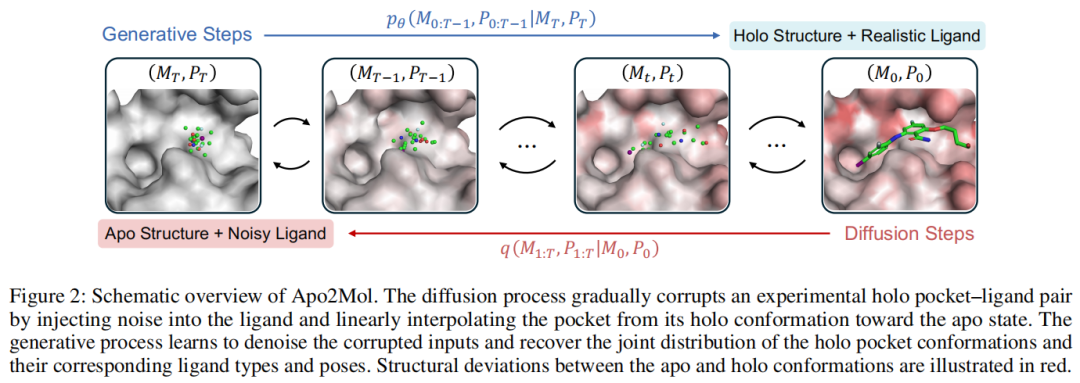
5. 方法细节:Apo2Mol 到底怎么做?
5.1 任务定义与输入输出
论文把任务定义为:
给定 apo 蛋白口袋 PA,生成对应的 holo 蛋白口袋 PH 和小分子 M。
其中:
- • PA 表示未结合态口袋,包含口袋原子坐标与原子特征;
- • PH 表示配体结合后的口袋构象;
- • M 表示生成的小分子,包含原子类型与 3D 坐标;
- • 学习目标是条件分布 p(PH, M | PA)。
对 ligand,模型直接预测原子类型和原子坐标。对 protein pocket,模型没有直接预测所有蛋白原子坐标,而是预测残基层面的三类构象变化:
- 1. translation: 残基局部平移;
- 2. rotation: 残基局部旋转,用四元数表示;
- 3. χ angle update: 侧链扭转角更新。
这个选择非常关键。蛋白不是自由点云。残基内部几何、主链连接、侧链局部结构都有强约束。用残基刚体变换和 χ 角更新来描述 pocket refinement,比直接逐原子生成更容易保持蛋白结构合理性。
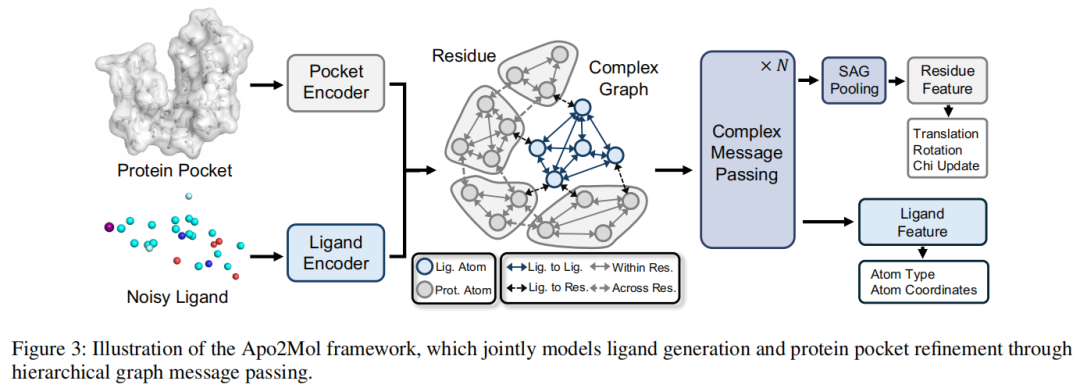
5.2 数据准备:从实验 apo-holo 对中构造监督信号
Apo2Mol 的训练样本由 apo pocket、holo pocket 和 holo ligand 组成。
具体处理流程大致如下:
- 1. apo-holo 结构对齐。 先用 RMSD alignment 将 apo 与 holo pocket 放到同一坐标系。
- 2. 残基层面变换计算。 使用 Kabsch 算法计算每个残基从 holo backbone 到 apo backbone 的平移与旋转。这里对齐的是残基主链 N-Cα-C 原子,并以 Cα 为局部中心。
- 3. 侧链 χ 角差异计算。 使用 DynamicBind 相关工具提取 apo 与 holo 的 χ 角,并计算 χ angle update。
- 4. 口袋定义。 holo pocket 被定义为 ligand 原子 10 Å 内的所有残基;对应 apo pocket 则从未结合结构中取相同残基。
这一步决定了模型学到的不是泛泛的蛋白构象变化,而是 ligand binding site 周围的局部结合诱导变化。优势是任务更聚焦;局限是 benchmark 中口袋定义依赖 holo ligand,而真实 apo-only 应用中往往需要先预测或指定口袋范围。
5.3 前向过程:同时破坏 ligand,并把 holo pocket 推回 apo pocket
Apo2Mol 的扩散过程比较有特点:小分子和蛋白口袋不是用同一种噪声处理。
对 ligand:
- • 坐标加入高斯噪声;
- • 原子类型进行 categorical corruption;
- • 随着时间步增加,clean ligand 逐渐变成 noisy ligand。
这与 TargetDiff、IPDiff 等 3D 分子扩散方法类似。模型最终要从 noisy ligand 恢复出原子坐标和类型。
对 protein pocket:
- • 终点不是标准高斯噪声,而是 apo state;
- • 模型从 holo pocket 出发,通过插值逐步得到接近 apo 的中间构象;
- • translation 和 χ angle 采用带噪声的线性插值;
- • rotation 使用四元数球面线性插值 Slerp,从 holo-to-apo rotation 平滑过渡到单位四元数,并加入小扰动。
直观理解:训练时,作者构造了一个从 holo complex 退回 apo pocket + noisy ligand 的路径。反向生成时,模型从 apo pocket + noisy ligand 出发,逐步恢复到 holo-like pocket + realistic ligand。论文图 2 展示了这个双向过程:前向过程将实验 holo pocket-ligand pair 腐蚀为 apo 结构和噪声 ligand,反向过程学习恢复 holo pocket 与 ligand 类型、pose 的联合分布。
这就是 Apo2Mol 与普通 3D ligand diffusion 的关键区别:它不是只对 ligand 加噪去噪,而是把 pocket refinement 也放入同一个扩散时间轴。
5.4 为什么用四元数?
蛋白口袋的局部旋转可以用旋转向量、欧拉角、旋转矩阵或四元数表示。Apo2Mol 选择四元数,原因是四元数在 3D 旋转插值中更稳定,可以避免欧拉角的万向节锁问题,也便于做平滑插值。
这在 apo-holo 转换中很重要。残基局部旋转不是简单的 3D 平移,如果旋转表示不稳定,模型可能在学习过程中产生不连续或数值不稳定的构象变化。论文消融也支持这一点:将四元数替换为旋转向量后,Vina min 和 QED 都下降。
5.5 网络结构:层级复合物图 + SE(3) 等变消息传递
Apo2Mol 的网络结构可以拆成四个部分。
1)蛋白与 ligand 分别编码
在每个扩散时间步 t,模型输入一个 noisy ligand 和一个中间状态 pocket。
- • ligand atom 使用原子类型 one-hot;
- • protein atom 使用三类 one-hot 特征:原子类型、氨基酸类型、是否 backbone atom;
- • 两者分别通过两层 MLP 投影到统一的 d 维隐空间。
2)构建 protein-ligand complex graph
编码后,Apo2Mol 基于空间邻近关系构建 kNN 复合物图。它不是把所有边都当成同一种边,而是定义四类边:
- 1. ligand 内部边;
- 2. ligand-residue 边;
- 3. 同一残基内部边;
- 4. 不同残基之间的边。
这个设计非常重要。因为 ligand 生成需要理解分子内部几何,pocket refinement 需要理解残基内部和残基之间的结构,ligand-pocket interaction 又需要跨分子信息传递。单一边类型会把这些不同物理关系混在一起,削弱模型对相互作用层级的表达。
3)SE(3) 等变注意力消息传递
复合物图进入 SE(3)-equivariant GNN。每个节点同时具有:
- • invariant feature h:表示化学语义、原子类型、残基环境等;
- • equivariant coordinate x:表示 3D 空间位置。
消息传递时,节点根据邻居距离、边类型、节点特征进行 attention-based aggregation,同时更新 h 和 x。这样做的意义是:如果整个复合物发生平移或旋转,模型输出也应以一致方式变化,不应因为坐标系选择不同而改变生成逻辑。
4)分别输出 ligand 与 pocket refinement
消息传递结束后:
- • ligand 的最终坐标直接来自最后一层 GNN 的坐标输出;
- • ligand atom type 由 ligand hidden feature 经过 MLP 分类得到;
- • protein pocket 先通过 SAGPooling 将 atom-level 表征聚合到 residue-level,再用多个 MLP head 分别预测 translation、rotation quaternion 和 χ angle update。
论文图 3 清楚展示了这一结构:protein pocket 与 noisy ligand 分别编码,构建包含多类边的 complex graph,经过多层 complex message passing 后,一路输出 ligand atom type 与 coordinates,另一路经过 SAGPooling 输出 residue-level pocket 更新。
5.6 训练目标:同时约束小分子与口袋变化
Apo2Mol 的总损失由五部分组成。
ligand 坐标损失
模型预测 clean ligand 坐标,与真实 ligand 坐标做均方误差。这个损失推动模型恢复正确的 3D pose。
ligand 原子类型损失
模型预测 ligand atom type,用 categorical distribution 的 KL divergence 进行监督。这个损失让模型学习不同原子类型在口袋环境中的空间分布。
pocket translation 损失
模型预测残基平移更新。目标不是简单的 apo-holo 差值,而是考虑旋转后的逆向 translation,使模型学习从中间状态恢复 holo 状态所需的几何变换。
pocket rotation 损失
rotation 使用 L1 损失,同时加入单位范数正则,确保预测四元数仍然是合法旋转表示。
pocket χ angle 损失
χ angle 是周期变量,直接用欧式差值会出问题。Apo2Mol 使用 cosine-based loss 处理角度差异,避免 359° 和 1° 被误认为相差很远。
最终损失是上述五项的加权和。这个训练目标体现了 Apo2Mol 的联合建模思路:ligand 的位置和类型、pocket 的平移、旋转与侧链角度都要同时被恢复。
5.7 推理流程:真实使用时怎么生成?
在推理阶段,用户给定 apo pocket。模型从随机 noisy ligand 开始,同时以 apo pocket 作为初始口袋条件,沿反向扩散过程迭代生成:
- 1. 当前 noisy ligand 与当前 pocket 构成 pseudo-interaction pair;
- 2. 复合物图网络预测 clean ligand 与 pocket 更新;
- 3. 根据扩散采样规则逐步减少 ligand 噪声;
- 4. 同时将 pocket 从 apo-like state 推向 holo-like refined state;
- 5. 最终输出一个 3D ligand 和一个 refined pocket conformation。
需要注意的是,论文正文对几个实际部署细节展开不多,例如 ligand 原子数如何确定、成键与化学清理如何完成、是否进行后续构象优化或 clash correction。这些细节对真实应用很关键,因为生成坐标和原子类型并不自动等于得到一个价态完全正确、可合成、可采购、可验证的小分子。
5.8 方法流程小结
可以把 Apo2Mol 总结成 7 步:
- 1. 从 PDB/PLINDER 中筛选实验 apo-holo-ligand triplets;
- 2. 对齐 apo 与 holo pocket;
- 3. 计算残基层面的 translation、quaternion rotation 和 χ angle update;
- 4. 在训练前向过程中,对 ligand 坐标和类型加噪,同时把 holo pocket 插值到 apo pocket;
- 5. 构建 noisy ligand + intermediate pocket 的层级复合物图;
- 6. 使用 SE(3) 等变 attention GNN 进行 ligand-pocket 消息传递;
- 7. 反向生成时,从 apo pocket 和 noisy ligand 出发,同时恢复 ligand 与 holo-like pocket。
6. 实验设计与关键结果
6.1 数据集与 benchmark
论文从 PLINDER 出发,构建实验解析的 apo-holo-ligand 数据集。筛选条件包括:
- • apo 与 holo 蛋白 100% 序列一致;
- • 结构分辨率 ≤ 2.5 Å;
- • 配体为药物样小分子;
- • 排除离子、辅因子、晶体 artifact、片段等;
- • 最终得到 24,601 个 triplets;
- • 时间划分:23,052 train、1,071 validation、478 test。
这个 benchmark 的亮点是大规模、实验结构来源、适合 apo-to-holo 学习。它比只在 holo pocket 上训练更贴近只有 apo 结构的早期靶点场景。
但它也有一个隐含问题:测试时的 pocket residues 是根据 holo ligand 10 Å 范围定义的,再映射到 apo 结构。这在 benchmark 中合理,但真实 apo-only 应用中,如果没有已知 ligand,就需要额外的口袋预测或活性位点定义流程。
6.2 对比方法
论文比较了四类代表性 SBDD 生成模型:
- • Pocket2Mol:基于口袋逐步放置原子;
- • TargetDiff:生成 ligand 原子坐标和类型,再后处理成键;
- • DecompDiff:将 ligand 分成 scaffold 和 arm,分别建模;
- • IPDiff:引入蛋白-配体相互作用先验的 3D 分子扩散模型。
FlexSBDD 和 DynamicFlow 因代码不可用未纳入比较。这个选择可以理解,但也削弱了与真正动态 pocket 方法之间的直接比较。
6.3 指标怎么看?
论文使用六类 ligand 指标:
- • Vina min: AutoDock Vina 打分,越低表示 docking score 越好;
- • QED: drug-likeness,越高越像已知药物性质空间;
- • SA: synthetic accessibility,越高表示合成可及性更好;
- • logP: 脂水分配系数,过低或过高都可能影响 ADMET;
- • Lipinski: 满足五规则的数量;
- • High Affinity: 生成 ligand docking score 优于参考 ligand 的比例。
此外,论文用 C-C bond distance 分布和 ground truth 的 Jensen-Shannon Divergence 评估分子几何合理性,用 apo-holo RMSD 分布与 apo-generated pocket RMSD 分布的 JSD 评估 pocket conformational change 是否接近实验分布。
这里需要强调:Vina score 是计算指标,不是实验亲和力。它适合初步比较生成结果,但不能证明真实活性。
6.4 apo 输入场景:Apo2Mol 在更现实设定下表现更好
第一组实验模拟真实场景:测试时只给 apo structure。baseline 模型虽然在 holo pocket 上训练,但测试时也输入 apo pocket;Apo2Mol 则从 apo pocket 生成 refined pocket 和 ligand,并在生成 pocket 上评估 ligand。
结果显示:
- • Apo2Mol 的平均 Vina min 为 -6.79,优于 IPDiff 的 -6.40;
- • 中位数 Vina min 为 -7.09,优于 IPDiff 的 -6.56;
- • High Affinity 达到 42.7%,高于 DecompDiff 的 34.3% 和 IPDiff 的 29.6%;
- • QED 平均值 0.59,也优于主要 baseline。
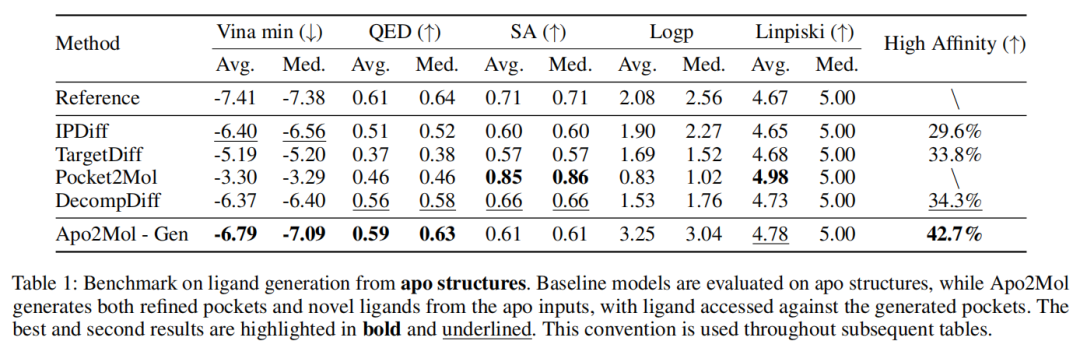
这个结果支持作者主张:当输入结构是 apo 时,显式建模 pocket refinement 有助于生成更适配的 ligand。
6.5 holo 评估场景:Apo2Mol 仍有竞争力
第二组实验中,baseline 在 native holo pocket 上评估,这是传统 SBDD 生成模型更有利的设定。Apo2Mol 仍然从 apo 输入生成 ligand,但 ligand 被拿到 native holo pocket 上评估。
结果显示:
- • Apo2Mol 平均 Vina min 为 -7.86;
- • IPDiff 为 -7.09;
- • Apo2Mol High Affinity 为 52.9%,IPDiff 为 44.9%;
- • Apo2Mol 的 QED 平均值达到 0.61,与 reference 接近。
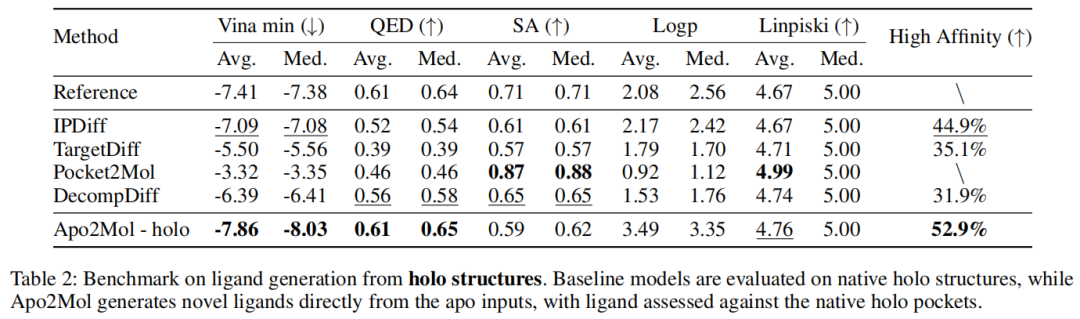
这说明 Apo2Mol 生成的 ligand 不只是适配自己生成的 pocket,在 native holo pocket 上也能保持较强 docking score。当然,这仍然是 docking-based evidence,不等价于真实结合自由能。
6.6 分子结构合理性:C-C 距离分布更接近真实分子
论文比较了生成分子与测试集真实分子的 C-C bond distance 分布。Apo2Mol 的 JSD 为 0.178,低于 IPDiff 的 0.216 和 TargetDiff 的 0.273。这个结果说明 Apo2Mol 生成的局部几何更接近真实分子分布。
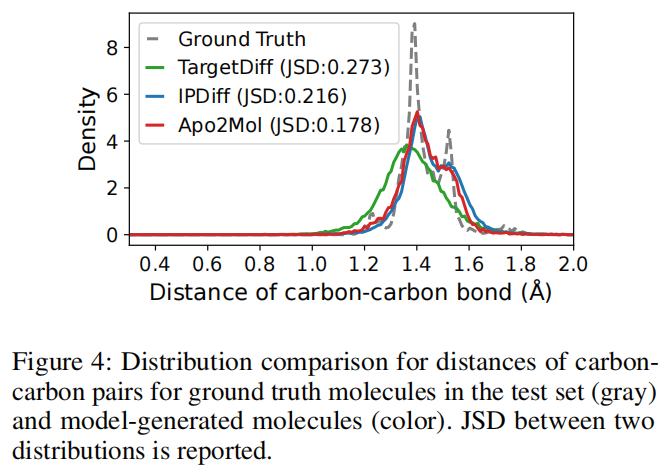
不过,C-C 距离只是很局部的几何检查。一个分子可以有合理的 C-C 距离,但仍可能存在价态错误、环张力异常、构象应变大、合成困难或不合理相互作用。因此这个结果可以支持结构 plausibility,但不能单独证明化学可用性。
6.7 pocket 结构合理性:能跟随 apo-holo 分布,但仍有差距
Apo2Mol 生成 pocket 与 apo structure 的 RMSD 分布,大体跟实验 apo-holo RMSD 分布一致,但 JSD 为 0.317,论文也承认存在 mild shift。这说明模型能够学到一部分结合诱导构象变化,但还没有完全捕捉不同蛋白、不同配体、不同局部环境下的精细构象多样性。
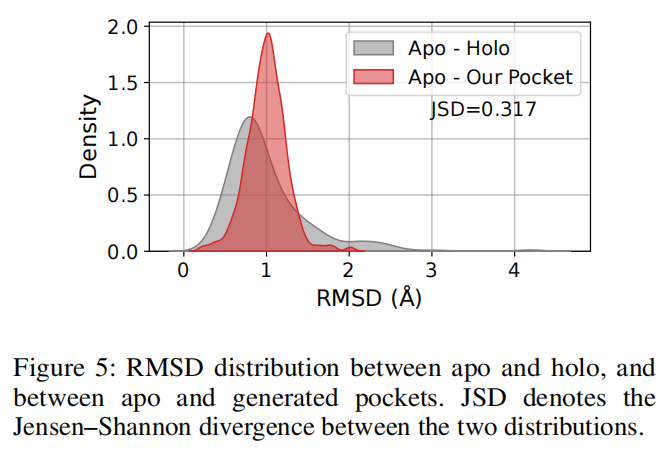
这也是 Apo2Mol 最重要的证据边界:它证明了联合生成 pocket conformation 是可行的,但还不能说明已经准确解决蛋白柔性问题。
6.8 消融实验:层级复合物图与四元数都重要
消融实验比较了完整模型、去掉 complex graph、去掉 quaternion 表示后的表现:
模型 | Vina min Avg | Vina min Med | QED Avg | QED Med |
|---|---|---|---|---|
Apo2Mol | -6.79 | -7.09 | 0.587 | 0.629 |
w/o complex graph | -6.18 | -6.22 | 0.524 | 0.529 |
w/o quaternion | -6.51 | -6.44 | 0.523 | 0.534 |
去掉 complex graph 后,模型不能充分区分 ligand 内部、ligand-pocket、残基内部和残基间相互作用,性能明显下降。去掉 quaternion 后,rotation 表示稳定性下降,也影响生成质量。这说明 Apo2Mol 的提升不只是来自数据规模,也与结构建模方式有关。
启发
启发一:分子生成不能只问 ligand 是否合理,还要问 pocket 是否处在可结合状态
很多 3D 分子生成论文关注 ligand 的 validity、QED、SA、Vina score,但忽略了口袋本身是否是适合结合的构象。Apo2Mol 把 pocket conformation 放进生成目标,提醒我们:SBDD 的条件不是静态背景,而是 ligand 设计的一部分。
启发二:apo-only 可能成为结构生成模型更现实的评估设定
真实项目中,holo 结构并不总是可得。只在 holo pocket 上评估生成模型,可能高估实际可用性。Apo2Mol 构建 apo-holo benchmark,并设置 apo 输入实验,是一个更接近早期药物发现的测试方向。
启发三:蛋白柔性不一定只能依赖 MD 数据
MD 轨迹提供连续过程,但成本高、偏差难控。Apo2Mol 使用实验 apo-holo endpoint 学习构象变化,为动态 SBDD 提供了另一种数据路线。未来更理想的方案可能是实验端点、MD 轨迹、AlphaFold ensemble、NMR ensemble、cryo-EM 多状态结构共同建模。
启发四:小分子生成正在走向复合物联合生成
早期模型生成 molecule;后续模型生成 molecule pose;现在开始生成 ligand-pocket complex。这个趋势非常重要,因为真实药物发现中的结构合理性本来就是复合物层面的性质,而不是单个分子层面的性质。
局限性
apo-holo 插值不是完整结合动力学
Apo2Mol 使用实验 apo 与 holo 端点,通过 translation、rotation 和 χ angle 插值构造中间状态。这是一种合理近似,但不等于真实结合路径。真实诱导契合可能存在多条路径、中间态、水网络重排、局部能垒、远端 allosteric 变化。线性或平滑插值能提供训练信号,却不能完全替代动力学采样。
pocket refinement 仍有分布差距
论文自己的 RMSD 分布结果显示,generated pocket 与真实 apo-holo 转换分布之间仍有 JSD = 0.317 的差距。这说明模型对平均趋势有学习能力,但对特定蛋白的细粒度 conformational transition 仍不充分。作者也在结论中指出,未来可通过更多蛋白结构预训练来缩小差距。
benchmark 口袋定义依赖 holo ligand
数据集中 holo pocket 定义为 ligand 10 Å 范围内残基,再提取 apo 中对应残基。这对监督学习是必要的,但实际 apo-only 项目中,用户往往没有参考 ligand,也不一定知道精确结合残基。真实部署需要结合 pocket detection、binding site prediction 或先验功能位点信息。
docking score 不是实验活性
Vina min 和 High Affinity 是重要但有限的指标。生成模型可能学会产生 docking score 较好的构型,但这些构型未必有真实结合、选择性、细胞活性或可合成性。尤其是当 pocket 也是模型生成的,score 可能同时受到 ligand 与 generated pocket 自洽性的影响。
化学有效性评价还不够完整
论文报告了 QED、SA、logP、Lipinski 和 C-C distance JSD,但真实药物发现还需要更多检查:
- • valence validity;
- • ring strain;
- • stereochemistry;
- • protonation state;
- • tautomer state;
- • PAINS/reactive group;
- • synthetic route availability;
- • novelty and diversity;
- • scaffold hopping 是否真实发生;
- • 与已知配体是否高度相似。
如果没有这些补充分析,模型生成的分子仍可能只是计算指标上看起来不错。
baseline 对动态方法比较不足
FlexSBDD 和 DynamicFlow 因实现不可用未加入 benchmark,这可以理解,但也意味着 Apo2Mol 与同样考虑 pocket flexibility 的最新方法缺少直接对比。此外,传统 flexible docking、ensemble docking、induced-fit docking 或基于多个实验构象的生成流程,也值得作为更贴近工业实践的参考。
没有实验验证
论文目前主要是计算验证。对 AI 制药来说,prospective synthesis and assay 仍是关键证据。Apo2Mol 是否能在真实靶点上产生可合成、可测、有效、选择性好的分子,还需要湿实验闭环来证明。
对 AI 制药未来发展的意义
Apo2Mol 的意义在于,它把一个经常被简化的问题重新放回模型内部:蛋白口袋不是静态容器,而是 ligand 设计过程中的动态参与者。
从未来发展看,这条路线可能推动几个方向。
基于动态结构生成
未来 SBDD 生成模型可能不再只输入一个 PDB pocket,而是输入一个结构集合、apo ensemble、AlphaFold ensemble 或 MD-derived conformational basin。模型输出也不只是单个 ligand,而是 ligand + binding pose + induced pocket + uncertainty。
flexible docking 与 de novo design 融合
传统 docking 主要解决已有 ligand 如何放入口袋,de novo generation 主要解决生成什么 ligand。Apo2Mol 这种方法把两者靠近:模型同时处理 ligand placement、ligand identity 和 pocket adaptation。未来可能出现生成-对接-局部重排-打分一体化模型。
从 endpoint 学习到路径学习
Apo2Mol 当前学习 apo/holo endpoint。下一步可以融合连续轨迹或多状态结构,让模型学习构象变化路径,而不仅是端点差异。这对处理大幅 loop movement、cryptic pocket、allosteric pocket、GPCR activation state 等问题会更重要。
评估体系需要升级
如果模型开始生成 pocket,那么评估也要从 ligand-only 指标升级到 complex-level 指标,例如:
- • pocket RMSD 与局部几何合理性;
- • side-chain rotamer correctness;
- • clash score;
- • hydrogen bond network recovery;
- • interaction fingerprint recovery;
- • generated pocket 的能量稳定性;
- • ligand 在 native holo、generated holo、MD relaxed holo 中的一致性;
- • prospective hit rate。
与闭环药物发现结合
Apo2Mol 适合作为候选生成器,但真实项目还需要后续闭环:
- 1. 生成 ligand-pocket complex;
- 2. 过滤化学有效性与合成可行性;
- 3. docking/MD/FEP 多层验证;
- 4. 合成与活性测试;
- 5. 将实验反馈用于模型再训练或 active learning。
只有走完这个闭环,动态口袋感知生成才可能从漂亮的 benchmark 结果转化为真实 hit discovery 能力。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-11,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号