微调 Boltz-2 实现更精准的先导化合物优化

微调 Boltz-2 实现更精准的先导化合物优化

DrugIntel
发布于 2026-06-01 17:11:21
发布于 2026-06-01 17:11:21

导读:本文深度解读2026年5月发表于bioRxiv的预印本论文《Affinity Fine-Tuning of Boltz-2: An Open Framework for Protein-Ligand Potency Prediction in Drug Discovery》。该工作由Biogen团队(Amini、Sciabola、Wang)完成,针对Boltz-2模型在实际先导优化项目中的落地痛点,提出了一套轻量级、可复现的项目特异性亲和力微调框架,并通过两项内部研究验证其有效性。
一、研究背景
在小分子药物研发流程中,先导化合物优化(Lead Optimization, LO) 阶段的核心目标之一是提升候选分子对靶蛋白的结合亲和力(Binding Affinity)。亲和力通常以 IC₅₀ 或 Kd 等形式表征,在药物发现中与成药性、选择性共同构成"三角优化"的核心。
准确的计算亲和力预测方法可以:
- • 在合成化合物之前筛选优先级,大幅降低实验成本
- • 指导构效关系(SAR)分析,加速化学空间探索
- • 替代或辅助高通量筛选,缩短研发周期
1.1 现有方法的局限性
当前主流计算方法可分为三类,各有明显的权衡:
方法类别 | 代表工具 | 精度 | 计算成本 | 可扩展性 |
|---|---|---|---|---|
物理自由能方法 | FEP+, Thermodynamic Integration | ★★★★★ | 极高(数天/分子) | 低 |
近似物理方法 | MM/GBSA, MM/PBSA | ★★★☆☆ | 中等 | 中 |
机器学习方法 | KDeep,DeltaDeltaG | ★★★☆☆ | 低 | 高,但泛化性受限 |
共折叠模型 | AlphaFold3, Boltz-2 | ★★★★☆ | 低–中 | 高(含结构信息) |
FEP+(自由能微扰,Free Energy Perturbation) 长期以来是先导优化中排序同系列配体的黄金标准。然而,其依赖分子动力学采样和精心构建的扰动图(perturbation map),导致:
- • 单分子计算耗时数小时至数天
- • 对蛋白构象灵活性(如P-loop柔性激酶)的处理依赖大量专家经验
- • 难以适应现代发现项目中每月数百个新合成化合物的通量需求
1.2 协同折叠模型的崛起
以AlphaFold3为开端,Boltz系列模型将蛋白与配体的结构预测统一在单一框架内,实现了联合结构-亲和力预测的范式转变:
- • Boltz-1(Wohlwend et al., 2024):首个开源协同折叠模型,实现生物分子相互作用的民主化建模
- • Boltz-2(Passaro et al., 2025):在Boltz-1基础上增加专用亲和力预测头,在公开FEP+基准上已接近FEP+精度,计算速度快数个数量级
然而,Boltz-2原始论文未公开训练方案(training recipe),导致研究者无法利用项目内部不断积累的实验测量数据对模型进行定制化适配——这恰恰是先导优化阶段最宝贵的信息资产。
二、核心问题:泛化模型 vs. 项目特异性需求
论文清晰地指出了一个领域内公认但未被充分解决的矛盾:
任何基于公开数据训练的ML亲和力模型,在应用于真实药物发现项目时,均面临严重的分布外(out-of-distribution)泛化问题。
具体体现在三个维度:
- 1. 靶点特异性:公开数据中特定靶点的覆盖极不均匀,稀有靶点或新型靶点的表示严重不足
- 2. 化学系列特异性:先导优化通常聚焦在结构高度相似的同系列(congeneric series)化合物上,其局部SAR往往编码了公开模型无法学习的项目专属信息
- 3. 测定条件特异性:内部生物测定的条件(缓冲液、温度、细胞系等)与公开数据存在系统性偏差,导致绝对pIC₅₀值存在不可忽视的偏移
论文中的实验数据有力佐证了这一问题:KDEEP默认版本在5个基准数据集中有3个出现负Pearson相关(R < 0),意味着在这些靶点上直接使用公开模型会产生误导性排序。
三、方法论:微调框架的四大技术组件
本工作的核心贡献是在公开Boltz-2代码库上构建了一套完整的微调基础设施,包含四个相互配合的技术模块。
3.1 Huber回归损失函数
模型对连续亲和力值(pIC₅₀)进行回归预测,采用Huber损失替代均方误差(MSE):
其中 ,以pIC₅₀单位计(即半个数量级)。
选择Huber损失的动机:内部测定数据存在典型的重尾分布(heavy-tailed distribution)——少数化合物因合成错误、测定失败或真实的非线性效应而产生异常值。MSE对这些异常值极敏感(损失与残差平方成比例),而Huber损失在大残差时退化为线性惩罚,有效抑制异常值对梯度的主导效应。
此外,论文还实现了成对排序损失(pairwise ranking loss) 和焦点二元分类损失(focal binary classification loss),但在所有报告实验中权重设为零,仅使用连续回归损失,为后续工作留下了扩展空间。
3.2 参数冻结策略
这是实现高效微调的关键设计决策。Boltz-2整体参数量约为515M,全量微调不仅计算成本高,在小数据集上还极易过拟合。
框架通过affinity_only_finetuning标志,将梯度更新严格限制在三个亲和力相关模块:
- •
affinity_module - •
affinity_module1 - •
affinity_module2
可训练参数从515M压缩至8.7M(约1.7%),同时所有冻结模块切换至.eval()模式,禁用dropout等随机操作,确保推理过程的确定性。
这一设计的理论依据是:Boltz-2的主干网络(trunk)在大量公开蛋白-配体数据上已学习到丰富的结构表示,这些通用表示可以作为固定特征提取器迁移到新靶点,而亲和力头则负责学习如何将这些表示映射到项目特定的pIC₅₀空间。
3.3 预计算嵌入缓存流水线
核心挑战:在参数全部冻结的情况下,如果每次微调迭代都重新通过整个Boltz-2主干进行前向传播,大量计算是冗余的。
解决方案:两阶段工作流
阶段一(预处理):将完整Boltz-2模型对整个数据集运行一次,缓存每个样本的单表示(single representations) 和对表示(pair representations)。
阶段二(微调):直接从磁盘加载缓存的表示,跳过冻结的主干和结构模块,仅执行亲和力头的前向和反向传播。
一个关键的工程细节:亲和力微调使用以配体为中心的裁剪(ligand-centered cropping),因此缓存的对表示必须重映射到裁剪后的token子集。框架通过在预处理阶段跟踪原始token索引,在微调时对预计算的pair张量进行重新索引来解决这一问题。
这一设计将微调的计算瓶颈从O(N × T_trunk)降低至O(N × T_head),使单GPU数小时内完成项目级适配成为可能。
3.4 亲和力感知数据处理
每条训练记录被封装为AffinityInfo对象,包含:
- • 连续亲和力值:log₁₀(IC₅₀),以µM为单位测定后转换
- • 二元结合标签:用于未来成对训练(当前实验中未使用)
- • 测定标识符(assay ID):为跨测定成对训练预留接口
- • 分子量:用于潜在的配体效率归一化
注释从YAML输入文件解析,并通过特征化器(featurizer)传播到每个样本的真实标签,实现从原始实验数据到模型输入的端到端流水线。
四、实验设计与结果分析
4.1 研究一:多靶点回顾性基准
数据集设置
采用Bansal et al.(2024)发布的基准数据集:
- • 172个化合物,分布在4个内部蛋白靶点、5个同系列化合物系列
- • 靶点构成:1个酶靶点(T1,含大型疏水口袋和多结合模式)+ 3个激酶靶点(T2有P-loop柔性,T3、T4)
- • T2分为两个独立数据集(T2-D1、T2-D2),反映不同药化优化阶段
- • 每靶点训练集规模:52–195个化合物
- • 评估指标:预测亲和力与实验值的Pearson相关系数R
比较方法
方法 | 类别 | 说明 |
|---|---|---|
FEP+ | 物理方法 | 原始基准中最强基线 |
DeltaDeltaG | ML方法 | 专为同系列成对差异学习设计 |
KDEEP(默认) | ML方法 | 基于3D卷积网络,开箱即用 |
KDEEP(微调) | ML方法 | 同等项目数据重训练 |
Boltz-2(默认) | 共折叠 | 公开检查点,无适配 |
Boltz-2(微调) | 共折叠 | 本文方法 |
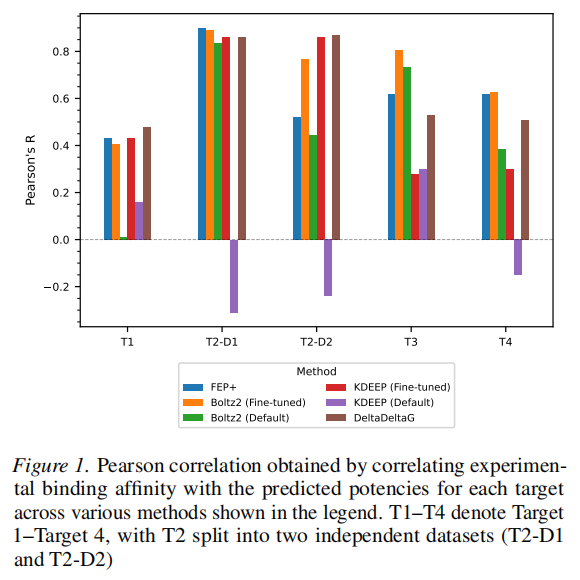
结果亮点
- • Boltz-2(微调)在5个数据集中的4个达到或超越FEP+(T2-D1、T2-D2、T3、T4)
- • 在T2-D2(P-loop柔性激酶),微调Boltz-2超越FEP+,提示结构型微调可隐式捕捉物理采样困难的构象效应
- • 在T1(最具挑战性的酶靶点),默认Boltz-2几乎无相关性(R ≈ 0.01),微调后恢复至R ≈ 0.40,接近FEP+(R ≈ 0.43)
- • 相比之下,KDEEP默认版在3/5数据集出现负相关,反映公开ML模型的严重泛化失败
4.2 研究二:单靶点时间序列评估
实验设计的特殊价值
本研究的设计极为贴近真实药物研发场景:
- • 化合物按月度批次合成和测定,时间跨度约两年
- • 定义19个时间切片(t5–t23),每个切片 使用前 批次数据训练,第 批次作为测试集
- • 训练集规模从约276个(早期切片)增长至1,714个(后期切片),共约1,734个配体
- • 模型必须泛化到训练截止后合成的结构新颖化合物,这是先导优化中最重要的预测场景
与真实场景的对应关系
这种时间序列评估规避了常见的数据泄露问题(如随机划分导致训练集和测试集结构相似),真实反映了模型在"预测未来"场景下的实用价值。
量化结果
方法 | 19切片均值R | 说明 |
|---|---|---|
Boltz-2(微调) | 0.76 | 本文方法,稳定领先 |
Boltz-2(默认) | 0.38 | 基础迁移信号,全程平稳但偏低 |
KDEEP(微调) | 与Boltz-2(微调)相近 | 在2个切片超越 |
Docking(Glide SP) | 静态基线 | 多数切片被微调Boltz-2超越 |
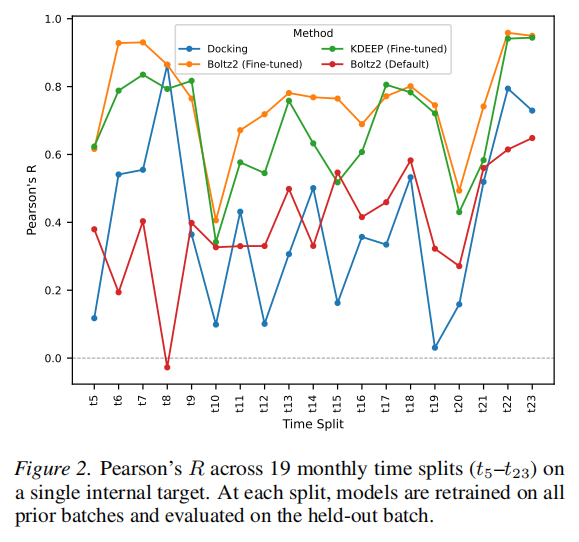
关键观察:
- • 早期切片(少量数据)下提升已十分显著,表明亲和力头在主干表示固定的情况下可高效适应有限数据
- • 中后期切片增益持续增大,体现了数据积累效应
- • 默认Boltz-2全程维持约0.38的相关性,说明公开模型对该靶点存在一定但不足的迁移能力
五、深度分析:为什么微调有效?
5.1 预训练表示的可迁移性
Boltz-2主干在大量公开蛋白-配体复合物数据上训练,学习到了关于分子间相互作用的丰富几何和化学表示。这些表示编码了:
- • 氢键、疏水相互作用、静电相互作用的空间分布模式
- • 蛋白结合口袋的几何特征
- • 配体的3D形状和药效团特征
这些通用特征对大多数靶点都有价值,因此可以作为高质量的固定特征提取器。
5.2 亲和力头学习的是什么?
微调过程中,亲和力头学习到的是:
- • 测定校准(assay calibration):将通用结合信号映射到项目特定的pIC₅₀量表
- • 靶点特异性加权:对于特定靶点,哪些相互作用特征更重要
- • 化学系列局部SAR:在高度相似的化合物系列中,细微结构差异如何影响亲和力
这种信息在公开数据中几乎不存在,只能从内部测定数据中学习。
5.3 为什么仅8.7M参数足够?
这一结果表面上令人惊讶,实际有充分理论支撑:信息瓶颈原理。
在特征提取质量足够高的情况下,从特征到标签的映射关系(亲和力头的任务)本质上是一个相对低维的问题。过多的参数反而会导致在小数据集上过拟合。8.7M的规模恰好在表达能力和泛化之间取得平衡。
5.4 对T1挑战性靶点的分析
T1是一个含大型疏水口袋和多结合模式的酶靶点,是原始基准中所有方法表现最差的靶点。微调后Boltz-2(R ≈ 0.40)相对FEP+(R ≈ 0.43)仍有小幅差距。
论文指出,这一差距可能反映的是结合位点本身的难度而非微调方法的局限——T1在原始基准中即是挑战最大的靶点,所有方法均未能展现突出性能。未来方向可能包括引入多结合模式感知的亲和力头设计。
六、局限性与未来方向
7.1 当前局限性
- • 内部数据集:两项研究均使用Biogen内部数据,无法完全独立复现;评估结果依赖于特定靶点和测定条件
- • 测试靶点数量有限:4个靶点的基准覆盖范围有限,不同靶点家族(GPCR、离子通道、蛋白-蛋白相互作用等)的泛化性有待验证
- • 成对损失未使用:论文实现了成对排序损失但未在实验中启用,可能低估了方法的潜力上限
- • 冷启动场景:早期切片(<50个化合物)下的性能未被详细分析
7.2 潜在扩展方向
- • 多测定联合微调:利用assay ID实现跨测定的迁移和校准
- • 主动学习集成:将微调模型的不确定性估计用于指导下一批合成优先级
- • 成对排序损失激活:在拥有足够数据的项目中启用,可能进一步提升排序性能
- • 不确定性量化:为预测提供置信区间,对先导优化决策更有实用价值
七、实践意义:如何在药物发现中应用?
对于有意将此框架应用于实际项目的研究者,论文隐含了以下实践指导:
数据准备
- • 至少准备50个化合物的pIC₅₀测定数据即可启动微调
- • 数据质量比数量更重要——异常值处理(Huber损失已部分缓解)和测定一致性至关重要
- • 建议使用与训练数据具有代表性化学多样性的测试集评估微调效果
计算资源
- • 单张现代GPU(A100/H100)可在数小时内完成微调
- • 预计算嵌入缓存是最耗内存的步骤,需为每个样本存储single和pair表示
部署建议
- • 随项目进展持续积累数据并定期重新微调(类似Study 2的时间序列设置)
- • 与FEP+互补使用:用微调Boltz-2快速筛选优先级,对顶级候选分子再用FEP+精确验证
八、小结
这项工作为基于结构的模型在特定化学项目中的适配提供了一种范式——不是重新训练整个基础模型,而是通过轻量级的任务头微调来利用内部实验数据。随着Boltz系列模型持续演进,这一框架的价值将随之放大。
但是值得注意的是微调Boltz-2 和 微调KDEEP性能并无太大差异,故 路漫漫,道阻且长~
参考信息
- • 论文标题:Affinity Fine-Tuning of Boltz-2: An Open Framework for Protein-Ligand Potency Prediction in Drug Discovery
- • 作者:Samad Amini, Simone Sciabola, Ye Wang(Biogen, Cambridge, MA)
- • 预印本日期:2026年5月26日
- • DOI:https://doi.org/10.64898/2026.05.26.727958
- • 开源代码:https://github.com/molecularinformatics/Boltz2_affinity
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-29,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号