计算表型药物发现:方法、挑战与未来方向

计算表型药物发现:方法、挑战与未来方向

DrugIntel
发布于 2026-05-08 19:23:59
发布于 2026-05-08 19:23:59

文献来源: Kumar S, Pal A & Chatterjee S. Unlocking the potential of computational phenotypic drug discovery: methods, challenges, and future directions. npj Systems Biology and Applications, 2026, 12: 46. https://doi.org/10.1038/s41540-026-00676-5
导读
表型药物发现(Phenotypic Drug Discovery, PDD)通过直接观察化合物对活体系统的效应来筛选候选药物,无需预先明确分子靶点。随着高通量生物数据的爆发式增长与机器学习技术的深度渗透,PDD 正从传统的经验性筛选演变为系统化、数据驱动的科学范式。本综述系统梳理了计算 PDD 的表型表征方法、核心算法工具、公共数据资源,深入剖析了制约其发展的主要瓶颈,并展望了若干极具潜力的前沿方向,为该领域研究者提供了结构化的方法论路线图。
一、背景:靶点药物发现的困境与 PDD 的复兴
1.1 现代药物研发管线的结构性挑战
现代药物研发管线高度结构化,涵盖靶点识别与验证、苗头化合物筛选、先导化合物优化、专利申报、临床前研究以及 I、II、III 期临床试验等阶段。这一过程资源消耗极为惊人——往往历经数年乃至十余年,花费数十亿美元,方能将一个候选药物推向市场。
尽管如此,药物研发的整体成功率依然偏低。核心矛盾在于:精准预测候选药物与生物靶点的相互作用,以及这种相互作用如何转化为真实的临床疗效,至今仍是生物医学科学面临的根本挑战。
1.2 靶点药物发现(TDD)的局限性
靶点药物发现(Target-Based Drug Discovery, TDD)遵循"机制优先"范式:首先阐明疾病的分子机制,继而针对特定靶点设计干预策略。AlphaFold 的出现使蛋白质结构预测精度大幅提升,虚拟筛选、分子动力学模拟、QSAR 建模等计算工具也使 TDD 流程显著提速。
然而,TDD 存在几个难以回避的内在局限:
- • 靶点-疾病关联的假设风险:大量通过 TDD 识别的分子靶点在转化到临床后未能产生预期疗效,揭示了分子层面的"击中靶点"与真实生理学效应之间的巨大鸿沟;
- • 功能表型捕获不足:聚焦孤立分子相互作用的方法难以充分反映药物在完整生命系统中的复杂功能输出;
- • 对未知生物学的盲区:对于致病机制尚未阐明的疾病,TDD 缺乏着力点。
1.3 PDD 的历史演变与当代复兴
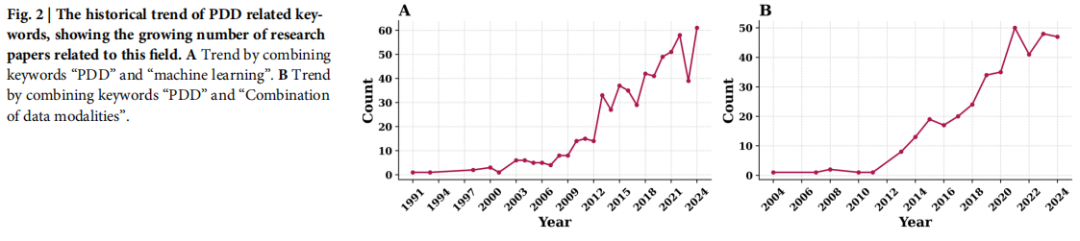
PDD 的根源可追溯至药理学的早期实践——通过经验性观察生物系统的表型变化来发现新治疗药物。20 世纪末,随着基因组学与分子生物学的兴起,TDD 一度占据主导地位。但近年来,高通量数据集的积累、成像技术的革命性进步以及人工智能的强势介入,使 PDD 重新成为制药科学的核心策略之一(见图:PDD 相关关键词发表趋势持续攀升)。
PDD 的核心优势在于其靶点无关性(target-agnostic):它直接评估化合物在细胞、组织乃至整体动物层面的生理效应,使全新作用机制的发现成为可能——这对于靶点未知的复杂疾病尤具价值。
1.4 PDD 标志性成功案例
药物 | 疾病领域 | PDD 发现路径 | 关键启示 |
|---|---|---|---|
Ivacaftor | 囊性纤维化 | 细胞表型筛选,发现时靶点未知 | CFTR 功能拯救先于机制阐明 |
Daclatasvir | 丙型肝炎 | HCV 复制子表型筛选 | 揭示了此前认为"不可成药"的 NS5A 蛋白 |
Risdiplam | 脊髓性肌萎缩症 | SMN2 剪接矫正表型筛选 | 开创 RNA 剪接靶向治疗新范式 |
Lenalidomide | 多发性骨髓瘤 | 表型筛选先于机制发现 | 其 cereblon 介导降解机制在上市后多年才被阐明 |
Thalidomide | 焦虑/多发性骨髓瘤 | 镇静表型发现,重定向用于血液肿瘤 | 靶点(cereblon)阐明远晚于临床使用 |
这些案例共同表明:PDD 具备发现"首创机制"(first-in-class mechanism)药物的独特能力,而这些机制在靶点驱动的框架下往往难以预见。
二、PDD 的技术景观
2.1 表型的多层次内涵
表型(Phenotype)涵盖生命系统一切可观测的特征,从分子变化、细胞形态到组织结构、生理功能乃至行为特征,是基因型与环境因素复杂交互作用的综合输出。在 PDD 框架中,表型作为真实治疗效果的代理指标,具有多个维度:
(1)细胞表型
细胞表型是 PDD 最核心的研究对象,涵盖:
- • 细胞活力(Viability):化合物对细胞存活的影响;
- • 细胞形态(Morphology):核形态、细胞骨架组织、胞质颗粒分布等;
- • 细胞周期状态:分裂前期、中期、后期、末期等各阶段识别;
- • 细胞功能:特定信号通路的激活或抑制。
细胞全景绘制(Cell Painting) 是当前最具代表性的多路复用表型检测技术。该技术利用六种荧光染料标记细胞的不同区室(核、内质网、高尔基体、线粒体、肌动蛋白/微管/细胞核),单次实验可提取超过 1500 个形态学特征,为化合物构建极为丰富的"细胞表型指纹"。
(2)生物体表型
多种模式生物被用于 PDD 的全系统表型评估,包括:
- • Caenorhabditis elegans(秀丽隐杆线虫)
- • Saccharomyces cerevisiae(酿酒酵母)
- • Drosophila melanogaster(黑腹果蝇)
- • 斑马鱼(Danio rerio)
相较于细胞模型,体内模型能够整合多组织信号传导网络,更真实地反映药物的生物利用度与系统效应,生物活性化合物的临床转化潜力通常也更高。
2.2 公共数据集资源体系
综述系统整理了支撑计算 PDD 研究的主要公共数据集,按数据类型分为以下几类:
形态学表型数据集
数据集 | 规模 | 主要用途 |
|---|---|---|
BBBC(Broad Bioimage Benchmark Collection) | 500+ 研究,690,000 张图像 | 细胞分割、表型分类、图像分析基准 |
Recursion(RxRx)系列 | 220 万张图像,83 TB | CRISPR 基因敲除与化合物扰动高通量成像 |
IDR(Image Data Resource) | 134 项研究,14,022,723 张图像,412 TB | 高质量生物成像数据公共存储库 |
JUMP-CP(Cell Painting 数据集) | 116k 化学扰动,12k 基因扰动,7500 万单细胞图像 | 大规模分子-形态关联预测 |
CytoImageNet | 890,737 张图像,894 类 | 生物图像迁移学习预训练 |
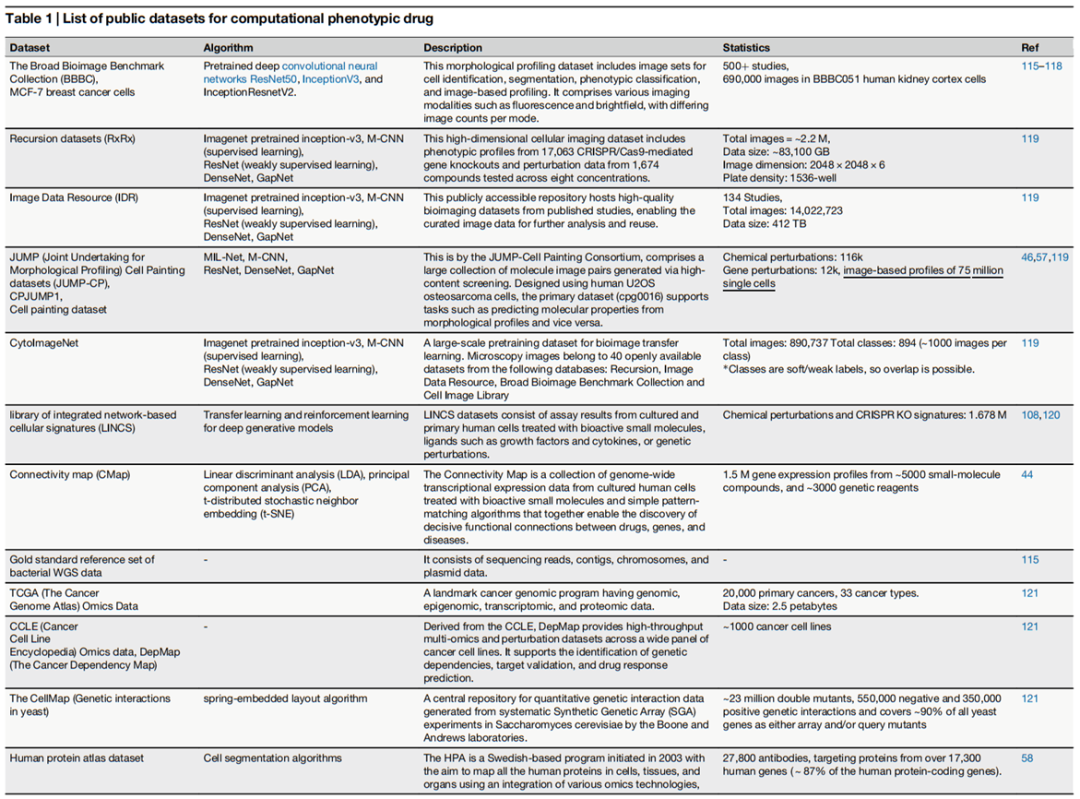
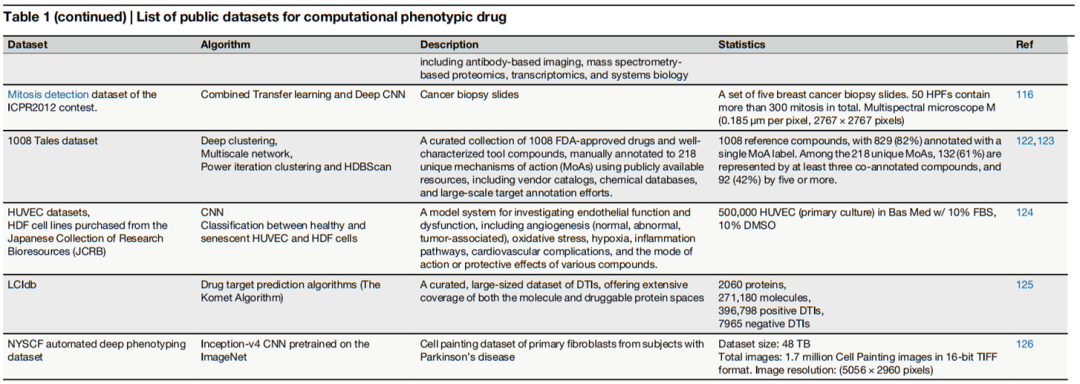
转录组与基因扰动数据集
数据集 | 规模 | 主要用途 |
|---|---|---|
LINCS L1000 | 1.678 M 化学扰动与 CRISPR KO 特征向量 | 药物转录组学特征,MoA 分析 |
CMap(Connectivity Map) | 1.5 M 全基因组表达谱,~5000 小分子 | 药物-基因-疾病功能连接发现 |
癌症与临床组学数据集
数据集 | 规模 | 主要用途 |
|---|---|---|
TCGA | 20,000 例原发癌症,33 种癌型,2.5 PB | 多组学癌症基因组研究 |
CCLE/DepMap | ~1000 癌细胞系 | 遗传依赖性、靶点验证、药物响应预测 |
Human Protein Atlas | 27,800 种抗体,覆盖 87% 人类蛋白编码基因 | 蛋白质组学图谱,细胞分割 |
参考化合物数据集
数据集 | 规模 | 主要用途 |
|---|---|---|
1008 Tales 数据集 | 1008 个 FDA 批准药物,218 种 MoA | MoA 分类与聚类基准 |
LCIdb | 2060 蛋白,271,180 分子,396,798 正例 DTI | 大规模药物-靶点相互作用预测 |
三、药物嵌入方法:从分子到向量
将化学结构与生物信息转化为机器学习可处理的数值表示(嵌入,Embedding),是计算 PDD 的基础性工作。综述归纳了四类主要嵌入策略:
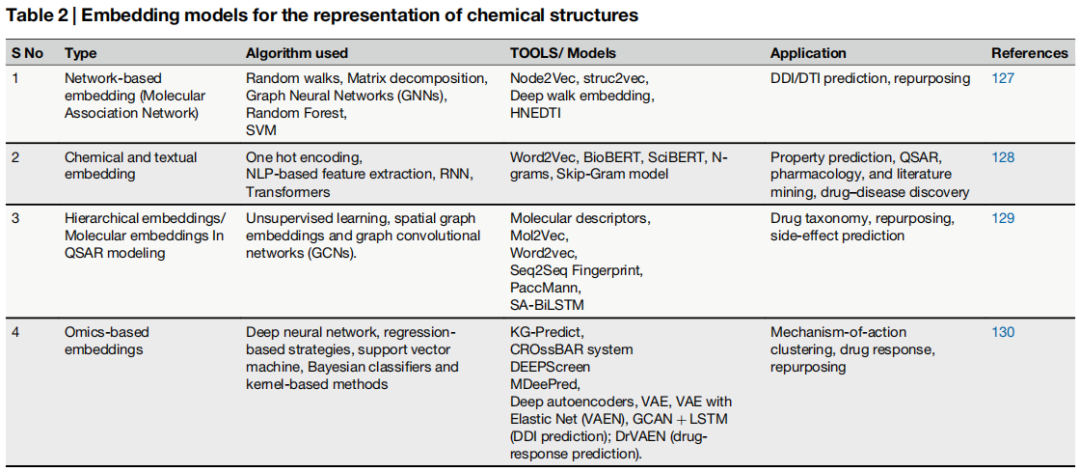
3.1 网络嵌入(Network-based Embedding)
核心思路:将药物及其相互关系表示为图(Graph),其中节点为药物/基因/蛋白/疾病,边为各类生物学关系(DDI、DTI、药物-疾病关联、化学相似性网络等)。
代表方法:
- • Random Walks(随机游走):Node2Vec、DeepWalk
- • 矩阵分解(Matrix Decomposition)
- • 图神经网络(GNN):HNEDTI 等
适用场景:药物-靶点相互作用(DTI)预测、药物重定向、副作用预测。
局限性:高度依赖已有领域知识,存在已知药物与靶点被过度代表、新型化合物被忽视的系统性偏差。
3.2 化学与文本嵌入(Chemical & Textual Embedding)
化学嵌入将分子结构编码为数值向量,包括:
- • 分子指纹(MACCS、featmorgan、Morgan 指纹)
- • SMILES 字符串表示
- • 基于深度学习的分子表示
适用于 QSAR 建模、相似性搜索和骨架跃迁(Scaffold Hopping),具有通用性强、可跨化合物库应用的优点。但其局限在于:纯化学结构不能反映生物系统中的分子间相互作用,结构相似性不等同于功能相似性。
文本嵌入基于自然语言处理,将科学文献转化为向量表示:
- • 词嵌入(Word2Vec)
- • 上下文嵌入:BioBERT、BERT、SciBERT、BlueBERT
- • 知识图谱嵌入
研究表明,将文献文本信息与化学结构嵌入结合,可显著提升药物重定向的预测性能。局限性主要包括:词共现不等于生物学相关性,计算资源需求大,以及难以捕获分子三维空间信息。
3.3 层级嵌入(Hierarchical Embedding)
将药物映射至双曲空间(Hyperbolic Space)中的树状几何结构,使功能或结构相似的药物在空间上相近。这一方法能够更精确地建模药物"分类学"——包括共享官能团、治疗靶点与代谢通路的层级关系。主要应用包括新适应症预测与不良反应预测。
该方法尚处于发展阶段,底层数学相对复杂,标准化最佳实践仍有待建立。
3.4 组学嵌入(Omics-based Embedding)
核心思路:利用深度学习模型从高维基因表达数据中提取降维的数值表示,解决传统组学分析中数据稀疏性、技术噪声、高维数据解读困难等问题。
代表工作:
- • Donner 等证明,基于学习转录组表示,具有相似表型效应的化合物可被有效聚类,验证了组学嵌入在 MoA 聚类与重定向机会识别方面的潜力;
- • Wang 等(GCAN + LSTM):将 LINCS L1000 数据的图卷积自编码器网络与 LSTM 结合,成功预测药物-药物相互作用(DDI),包括磺脲类药物联用致低血糖及二甲双胍相关乳酸酸中毒等临床关键问题。
局限性:高质量组学数据对多数药物而言仍不可及;批次效应(Batch Effect)影响重复性;计算资源与专业技术要求极高;存在过拟合风险。
四、预测模型体系
4.1 基于特征的传统机器学习模型
这类模型以结构化特征(分子描述符、理化性质、高通量筛选结果、基因表达谱)为输入,通过监督学习建立特征与生物效应之间的映射关系。
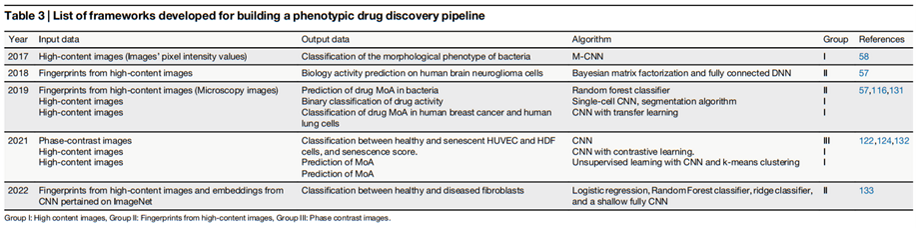
代表算法及应用:
- • 支持向量机(SVM):Zhao 等人证明其在跨多样化学数据集的毒性活性预测方面具有较强泛化能力;
- • 随机森林(Random Forest)与梯度提升机(XGBoost):对转录组等高维数据集尤为有效;
- • 整合平台:RepurposeDrugs、DrugRepo 等整合结构相似性、通路数据与疾病-基因关联,实现跨数百种疾病的重定向预测。
优势:计算效率高,可解释性强。局限:高度依赖已有注释,对发现全新作用机制的化合物效果有限。
4.2 深度学习模型
深度学习从大规模数据中自动学习生物学意义的特征表示,在毒性预测、药物响应预测、分子生成等任务上表现出色。
代表模型与应用:
模型 | 核心方法 | 主要应用 |
|---|---|---|
DrVAEN | 变分自编码器(VAE)+ 弹性网络,嵌入治疗前转录组 | 患者个性化药物响应预测与生存结局预测 |
GCAN + LSTM | 图卷积自编码器 + 长短时记忆网络 | DDI 预测,包括临床相关相互作用 |
MolDiffusion | 扩散模型(Diffusion Model) | 以目标表型特征为条件生成新分子 |
scGen | 单细胞生成模型 | 预测细胞对扰动的响应,识别可逆转疾病状态的化合物 |
cGAN-Cell Painting | 条件生成对抗网络 + 形态学图像 | 生成与目标视觉细胞效应匹配的分子 |
PAE/PertVAE | 扰动自编码器 | 提取细胞扰动核心特征,生成或对抗特定生物效应 |
4.3 网络基模型
网络基模型将生物学编码为关系网络,建模基因、蛋白质、药物、疾病之间的动态相互作用,其核心假设是:生物功能与药物响应涌现于分子网络的整体互作,而非单个分子的孤立行为。
代表工具:
- • MMAtt-DTA、DTiGEMS+、DGraphDTA:利用 GNN 对药物分子图与蛋白质图分别建模,实现深度药物-靶点亲和力预测;
- • PGxDB(药物基因组学平台):整合分子靶点、不良反应及适应症数据,支持个性化医学研究。
4.4 单细胞与多组学整合模型
这一前沿方向利用单细胞测序数据,在单细胞分辨率下预测药物效应:
- • DrugReSC:将疾病细胞的"指纹"与已知药物效应库比对,精准识别能将病态细胞逆转至正常态的候选药物;
- • Drug2Cell、scTherapy:识别药物影响最显著的细胞亚群,发现具有高效低毒特征的药物组合;
- • 多模态整合框架:融合化学结构、基因表达谱和蛋白质相互作用,构建多维药物表征,实现更可靠、更普适的预测。
4.5 可解释人工智能(XAI)在 PDD 中的应用
面对深度学习模型的"黑箱"问题,XAI 方法为 PDD 决策提供透明化依据:
- • SHAP(SHapley Additive ExPlanations):量化每个特征(如特定基因表达水平、化学子结构)对预测结果的贡献,同时提供全局(模型整体行为)与局部(单次预测)视角;
- • LIME(Local Interpretable Model-Agnostic Explanations):围绕单次预测构建局部代理模型,解释特定输出的成因。
XAI 在 PDD 中的典型应用包括:
- • 抗癌肽预测:t-SNE 可视化与 SHAP 分析揭示理化性质和组成特性对预测的决定性贡献;
- • 抗 MRSA 肽研究:RCEMT(能量估计)、ExPseAAC(理化性质)、DDE(序列衍生特征)等特征集的重要性量化;
- • 传统中医(TCM)多成分-多靶点-多通路体系的机制解码;
- • 药物 ADMET 性质的安全性相关特征识别;
- • 药物-药物与药物-靶点相互作用的机制解析。
战略价值:XAI 不仅提升模型可信度,更能辅助临床试验设计与患者分层,为监管审批提供可追溯的科学依据。
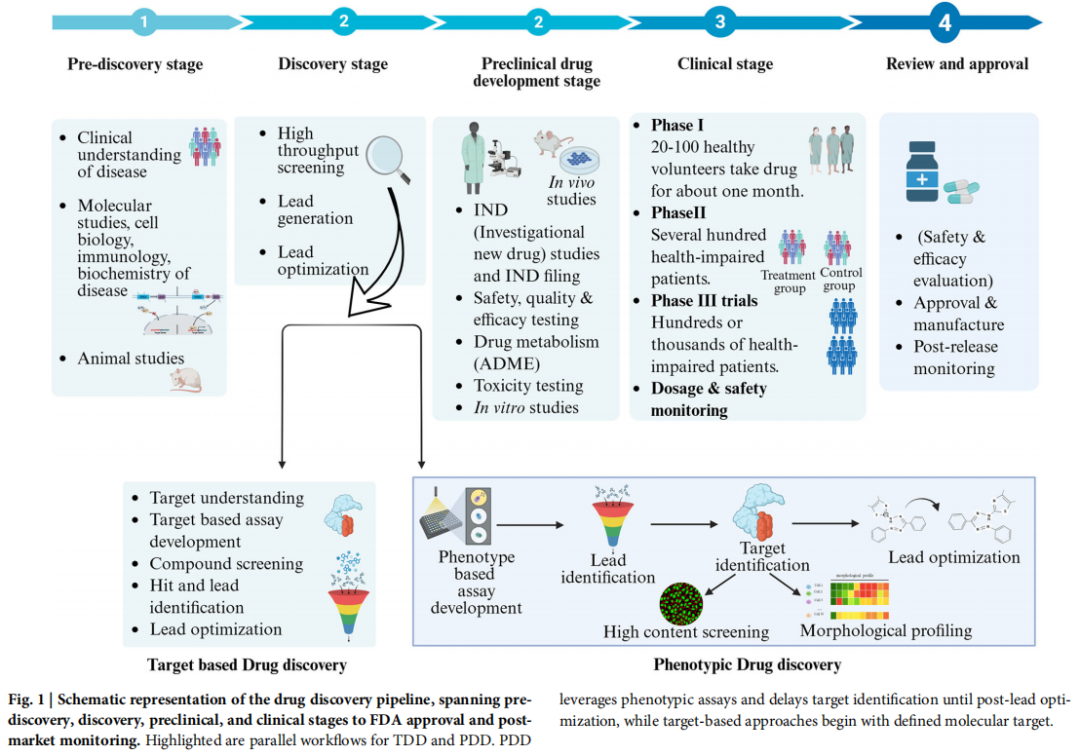
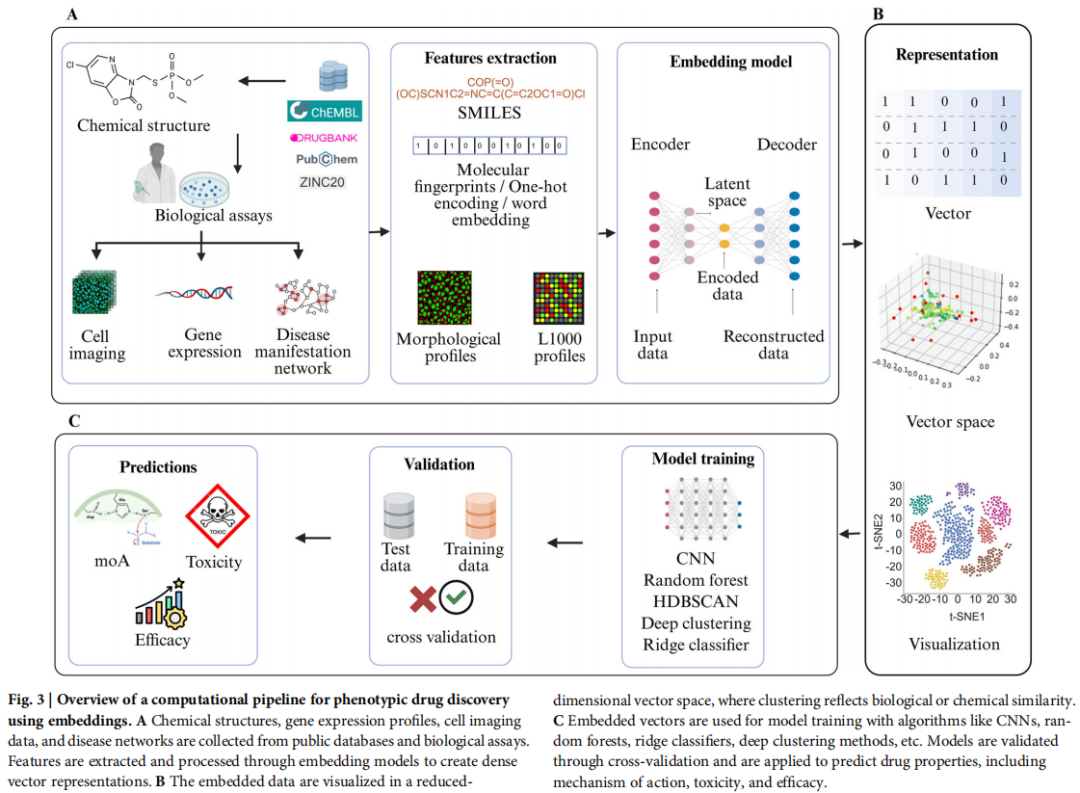
五、计算 PDD 流程总览
综述将完整的计算 PDD 流程概括为以下三个阶段(对应论文图 3):
┌─────────────────────────────────────────────────────────┐
│ 阶段 A:数据采集与嵌入表示 │
│ │
│ 输入源:化学结构数据库(ChEMBL、DrugBank、PubChem) │
│ 生物学测定数据(细胞成像、基因表达、疾病网络) │
│ │
│ 特征提取:SMILES → 分子指纹 / One-hot 编码 / 词嵌入 │
│ 形态学图像 → 形态学特征谱(Cell Painting) │
│ 基因表达 → L1000 特征谱 │
│ │
│ 嵌入模型:编码器-解码器架构 → 潜空间向量表示 │
└───────────────────┬─────────────────────────────────────┘
│
┌───────────────────▼─────────────────────────────────────┐
│ 阶段 B:降维可视化与相似性评估 │
│ │
│ 向量空间中生物学/化学相似性聚类 │
│ (t-SNE、UMAP、PCA 等降维可视化) │
└───────────────────┬─────────────────────────────────────┘
│
┌───────────────────▼─────────────────────────────────────┐
│ 阶段 C:模型训练、验证与预测 │
│ │
│ 算法:CNN、随机森林、HDBSCAN、深度聚类、岭分类器等 │
│ 验证:交叉验证(Cross-Validation) │
│ 预测输出:作用机制(MoA)、毒性、疗效 │
└─────────────────────────────────────────────────────────┘六、主要挑战:三大核心瓶颈
6.1 数据稀缺与标准化缺失
表型测定产生的数据种类繁多——从转录组学、蛋白质组学到形态学和功能性表型——但这些数据往往高度分散,跨平台和跨研究的整合面临严重障碍。数据标准化的缺失显著降低了 ML 模型的训练质量,导致预测性能与泛化能力不足。
典型失败案例:
- • Mylotarg:抗体-药物偶联物,因效率过低而撤市;
- • Gefitinib(Iressa):在广泛人群中未能证明生存获益,在 III 期临床前终止;
- • Fialuridine(抗病毒药)与 TAK-875(代谢疾病药):均因肝损伤而终止。
症结分析:现有公共数据集缺乏疾病模型特异性的丰富上下文信息,制约了稳健计算预测模型的开发。
6.2 表型筛选的验证瓶颈
不同于靶点药物筛选中定义明确的单一终点(如酶抑制率),表型筛选依赖多参数读出(细胞形态、行为、活力等),这使得自动化、解读和重复性均面临巨大挑战。
表型命中化合物需在从细胞测定到动物模型的不断复杂化的生物系统中逐级验证,而体内疾病模型在向人体转化时常遭遇方法论差异与物种特异性壁垒。
典型教训:
案例 | 失败原因 | 教训 |
|---|---|---|
DMXAA(黄酮类抗癌化合物) | 选择性激活鼠源 STING,而非人源 STING | 物种特异性机制差异的隐患 |
BIA 10-2474(FAAH 抑制剂) | 早期表型研究未进行严格的靶点解卷积,实际抑制多个脱靶脑丝氨酸水解酶,I 期临床致一人死亡 | 跳过严格靶点去卷积的灾难性后果 |
Thalidomide(沙利度胺) | 以镇静效果获批,数十年后才识别出 cereblon(CRBN)靶点,为时已晚地揭露了致畸机制 | 早期机制验证的关键重要性 |
NXY-059(神经保护候选药) | 啮齿类卒中模型效果显著,但未建立清晰 MoA,模型生物学相关性不足,大型临床试验完全失败 | 依赖非预测性表型模型的根本风险 |
6.3 靶点解卷积的机制模糊性
识别有效化合物只是 PDD 的第一步;阐明其作用机制(Mechanism of Action, MoA)才是真正的挑战。与 TDD 中机制预先确定不同,表型命中化合物往往需要生物学"侦探工作"才能解码其靶点。这一过程劳动密集、耗时漫长,且成功率有限。
挑战因多药理学(Polypharmacology,即化合物同时作用于多个靶点)的普遍存在而进一步加剧——单一靶点归因本身可能就是一种过度简化。
然而,机制模糊性也孕育了**药物重定向(Drug Repurposing)**的机遇:
药物 | 原始适应症 | 重定向新适应症 | 机制转变 |
|---|---|---|---|
Sildenafil | 心绞痛与高血压 | 勃起功能障碍 → 肺动脉高压(PAH) | PDE5 抑制的多组织效应 |
Imatinib | 慢性粒细胞白血病(CML,BCR-ABL 抑制) | 胃肠道间质瘤(GIST,KIT/CD117 抑制) | 激酶选择性的新发现 |
Thalidomide | 镇静 | 多发性骨髓瘤 | 免疫调节与 CRBN 介导降解 |
Minoxidil | 抗高血压 | 局部脱发治疗 | 血管扩张的局部转化应用 |
七、未来方向
7.1 iPSC 与 3D 类器官模型
诱导多能干细胞(iPSC)可以从患者皮肤或血液细胞重编程为任意细胞类型,提供个性化疾病模型平台。与 3D 类器官培养结合后,这些细胞能自组织形成微型组织结构,其生理行为远比传统二维培养更接近真实人体器官,为个性化表型筛选开辟新途径。
7.2 生成式 AI 与基础模型
生成式 AI 模型(如 MolDiffusion)能够以目标表型特征为条件,在化学空间中生成满足特定要求的新分子,显著扩展可探索的化学空间。基础模型(Foundation Models)具备从少量数据中学习的能力,对于数据稀缺的罕见病领域尤具价值。生成式 AI 还可模拟基因表达模式,以匹配个体层面的表型特征,推动精准医学落地。
7.3 单细胞分辨率的表型分析
单细胞测序技术的成熟使在单细胞分辨率下解析药物效应成为现实。DrugReSC、Drug2Cell、scTherapy 等工具已能精准定位药物影响最显著的细胞亚群,并识别高效低毒的协同药物组合,为系统性个体化治疗奠定基础。
7.4 先进成像与 AI 驱动的靶点解卷积
高分辨率成像技术与先进细胞图像分析算法的结合,使研究者能够将此前"观察有效但机制未知"的表型命中转化为可理解的生物学过程。化学探针(Chemical Probe)与基于化学生物学的靶点解卷积方法(如 DARTS、化学蛋白质组学、活性蛋白质谱)的系统整合,将是未来破解 MoA 谜题的关键技术路径。
7.5 多组学数据整合与 AI 协同
AI 与多组学技术(转录组学、蛋白质组学、磷酸化蛋白质组学等)的深度融合,正在增强细胞疾病表型识别的系统性与普适性。多模态框架——同时整合化学结构、基因组学影响、蛋白质互作网络——是提升预测可靠性与广度的必然路径。
八、方法论路线图:研究者实践指南
研究问题界定
│
▼
表型选择(细胞表型 / 生物体表型)
│
├── 细胞成像(Cell Painting 等)
├── 基因表达谱(L1000 / RNA-seq)
└── 蛋白质组 / 磷酸化蛋白质组
│
▼
数据预处理与特征提取
│
├── 形态学特征(CellProfiler 等)
└── 分子嵌入(SMILES / 图神经网络等)
│
▼
嵌入建模与相似性评估
│
├── 网络嵌入
├── 化学 / 文本嵌入
├── 层级嵌入
└── 组学嵌入
│
▼
预测模型训练与验证
│
├── MoA 预测
├── 毒性预测
├── 药物-靶点亲和力
└── 药物重定向
│
▼
可解释性分析(SHAP / LIME)
│
▼
实验验证(细胞模型 → 动物模型 → 临床转化)
│
▼
靶点解卷积(化学蛋白质组学 / 计算方法)写在最后
这篇综述的最大价值在于提供了一个统一的方法论框架,将表型表征、计算嵌入、预测建模与验证策略整合为一条完整的 PDD 研究路线图。几点深层启示值得关注:
- 1. PDD 与 TDD 并非对立而是互补:PDD 的"化合物优先"策略与 TDD 的"机制优先"方法在本质上是互补的,结合使用可显著提升发现新药的概率,特别是对于复杂的多靶点疾病。
- 2. 数据质量是根本瓶颈:无论算法多么先进,训练数据的质量、标准化程度与领域特异性,始终是 ML 模型预测性能的决定性因素。推动开放数据共享与数据标准化是整个领域亟需的系统性工程。
- 3. 机制验证不可或缺:BIA 10-2474 等案例的惨痛教训表明,在推进临床开发之前,严格的靶点解卷积与脱靶效应评估是必要的安全防线,而非可选项。
- 4. XAI 是连接模型与信任的桥梁:在监管日趋严格的背景下,可解释性不仅是学术追求,更是 AI 辅助药物发现走向实际应用的前提条件。
- 5. 生成式 AI 将重塑药物发现范式:从被动筛选到主动设计——生成式模型使"以目标表型为条件生成候选分子"成为可能,这将从根本上改变 PDD 的操作逻辑。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-05,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号