Nat. Comput. Sci. | 通用机器学习力场的实验测量基准评估

Nat. Comput. Sci. | 通用机器学习力场的实验测量基准评估

DrugAI
发布于 2026-07-17 20:59:11
发布于 2026-07-17 20:59:11
DRUGONE
通用机器学习力场有望以接近量子力学计算的精度和远低于密度泛函理论的成本,实现跨元素、跨材料体系的大规模原子模拟。然而,现有模型主要在计算生成的数据集上训练和测试,其评价指标通常集中于静态结构的能量和原子力误差,难以反映材料在实验条件下的真实表现。
研究人员提出 UniFFBench,并构建了实验驱动的 MinX 数据集。该数据集收录1500余种矿物体系,覆盖85种元素,温度范围为0至5000 K,压力最高达到1000 GPa,同时包含复杂多元素组成、部分原子占位、结构无序和实验弹性数据。研究人员在统一模拟协议下评估了 CHGNet、M3GNet、MACE、MatterSim、SevenNet 和 Orb 六种先进通用机器学习力场,并进一步使用 UMA 和 MACE-OMat 分析大规模训练数据对模型能力的影响。
结果显示,不同模型的分子动力学稳定性差异显著。Orb 和 MatterSim能够完成全部测试,而 CHGNet 和 M3GNet 在多数矿物体系中出现模拟失败。即使是稳定性最好的模型,其密度预测误差仍高于实际材料应用通常能够接受的范围。研究还发现,模拟稳定性与弹性性质准确性并不一致,能够稳定维持晶体结构的模型仍可能严重误判弹性张量。误差与训练数据中元素对和化学环境的出现频率密切相关,说明现有模型更接近训练化学空间内的插值工具,而非真正具有普适迁移能力的物理模型。

通用机器学习力场试图用神经网络学习材料的势能面,从而在远低于量子力学计算的成本下预测原子能量、受力、应力和运动轨迹。随着训练数据和模型规模扩大,这类方法已经能够覆盖周期表中的大部分元素,并被用于晶体结构弛豫、材料筛选、分子动力学模拟和新材料发现。
然而,当前机器学习力场大多依赖密度泛函理论数据进行训练。许多训练结构虽然来自实验晶体数据库,但能量、原子力和应力仍由密度泛函理论重新计算。模型随后又在相似的密度泛函理论数据上接受评价,形成了潜在的训练—评价闭环。模型可能只是高精度复现某种计算方法的结果,却未必能够准确对应实验中可测量的密度、晶格常数、热膨胀和弹性性质。
真实材料还包含标准计算数据集较少覆盖的复杂因素。例如,天然矿物可能含有十余种甚至二十余种元素,晶胞中包含数百个原子,并存在原子位点部分占位、成分无序、缺陷以及复杂配位环境。材料在高温或高压下还会发生显著的热振动、体积变化和非谐响应。仅根据零温静态结构的能量和原子力误差,很难判断模型是否能够在这些条件下维持稳定而合理的动力学轨迹。
因此,研究人员认为,通用机器学习力场需要接受独立于训练目标的实验检验。评价不仅应关注能量和原子力,还应考察模型能否维持晶体结构、再现实验密度和晶格参数、保持合理的原子空间分布,并准确预测材料的力学响应。
方法
研究人员从矿物晶体结构数据库和相关文献中整理实验结构,经过空间群符号统一、元数据校正、结构解析和质量控制后构建 MinX。该数据集包含四个互补子集:MinX-Eq 用于测试常温常压附近的平衡矿物结构;MinX-HTP 包含极端温度和压力条件下的体系;MinX-POcc 收录具有原子部分占位的矿物,并通过构建近似各向同性的超胞和按占位概率分配原子种类,将统计占位转换为可模拟的有序结构;MinX-EM 则包含100种具有实验弹性张量的矿物。研究人员将晶胞扩展至约100至200个原子,先进行结构最小化,再开展50 ps的恒温恒压分子动力学模拟。模型评价包括模拟能否完成、密度和晶格参数误差、径向分布函数、键长变化以及弹性张量、剪切模量和杨氏模量。模拟过程中若发生数值发散、键断裂、原子重叠,或结构性质误差超过100%,则被视为失败。研究人员还分析了不同元素对的键长误差、训练数据中的出现频率以及原子间相互作用曲线的平滑程度,以确定模型失效的来源。
结果
UniFFBench框架与MinX数据集
UniFFBench将模型、实验数据和多维评价指标整合到同一框架中。与常用训练数据集 MPtrj 相比,MinX在元素总覆盖范围上相近,但化学组成和结构复杂度明显更高。MPtrj中的单个结构最多含有9种元素,而 MinX中的矿物最多包含23种元素。MinX中还存在大量含数百个原子的晶胞,这些体系显著超出了多数训练配置的规模和复杂程度。
MinX-HTP进一步覆盖宽广的温度和压力区间,用于检验模型在远离常温常压条件时的稳定性。MinX-POcc则模拟天然材料中常见但在标准密度泛函理论数据集中经常被排除的部分占位和成分无序。通过这些子集,UniFFBench不仅比较模型的平均精度,也能够识别它们在特定化学环境和实验条件下的失效模式。
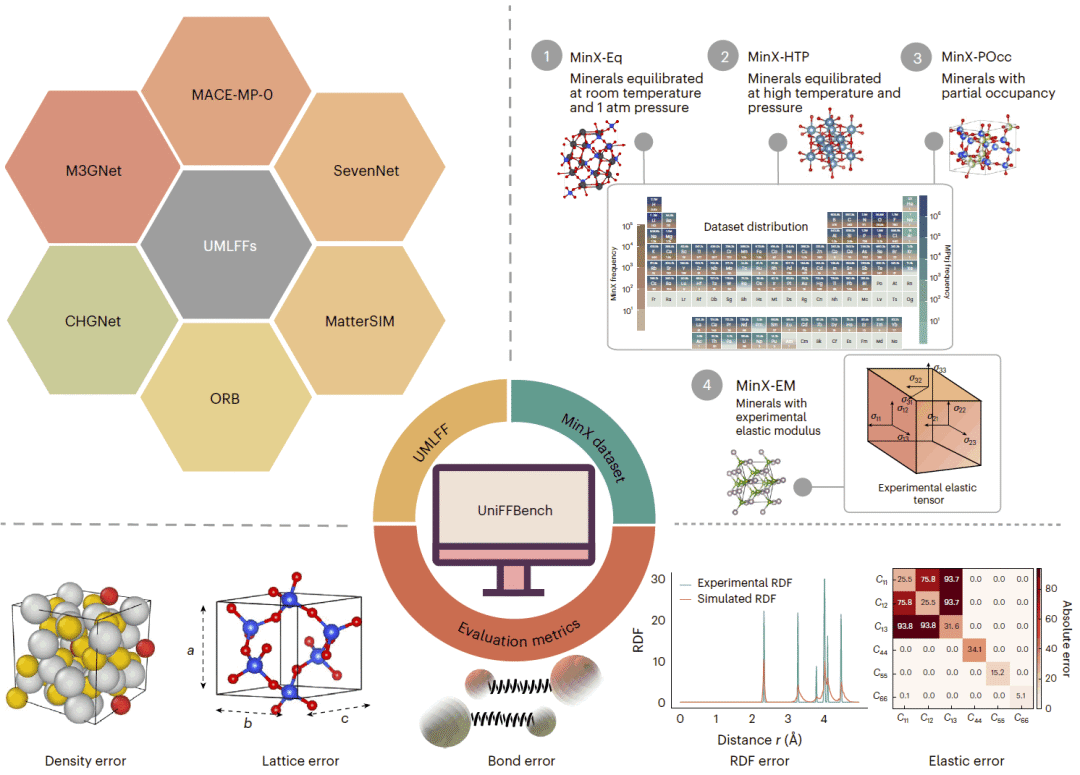
图1:UniFFBench整体框架,包括待评估模型、MinX四个子集以及结构、动力学和力学性质评价指标。
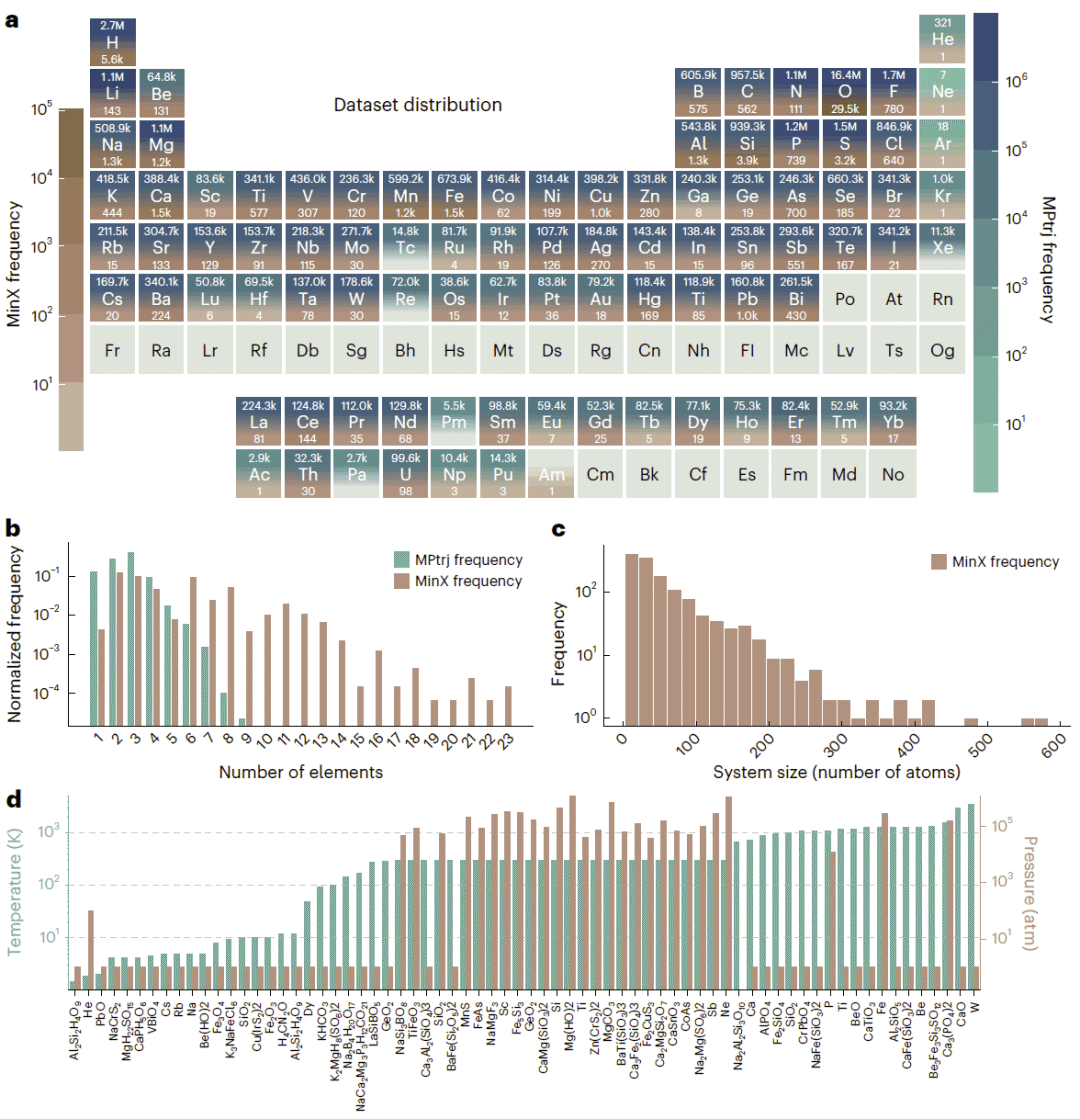
图2:MinX与常用训练数据集的元素分布、组成复杂度、晶胞规模以及温度和压力范围比较。
分子动力学模拟稳定性
六种模型在模拟稳定性方面呈现明显分层。Orb和MatterSim在三个主要子集中均实现了100%的轨迹完成率。MACE和SevenNet表现居中,在高温高压子集中的完成率约为95%,但在部分占位体系中下降至约75%至80%。这表明成分无序和复杂占位结构对模型的泛化能力构成了更严峻的挑战。
相比之下,CHGNet和M3GNet在各子集中均表现出超过85%的失败率。部分失败源于模型给出异常巨大的原子力,使数值积分无法继续;另一些模拟则出现键断裂、原子重叠或晶体结构快速塌缩。值得注意的是,模拟早期的能量和原子力误差与后续稳定性相关性很弱,因此较低的初始误差不能作为长时间动力学稳定的可靠保证。
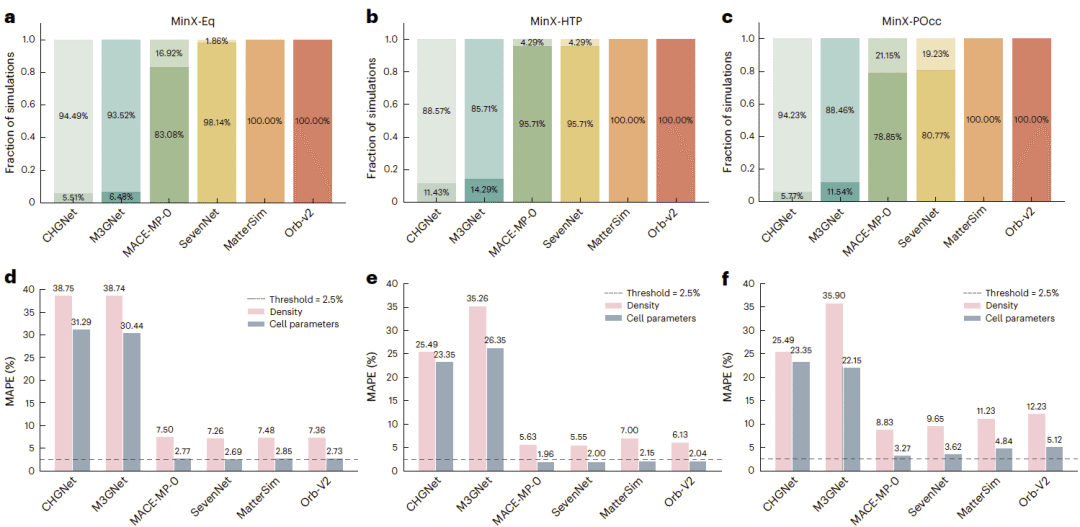
图3:不同模型在MinX-Eq、MinX-HTP和MinX-POcc上的模拟完成率,以及密度和晶格参数预测误差。
结构预测准确性与动态演化
在能够稳定完成模拟的模型中,Orb、MatterSim、SevenNet和MACE对密度和晶格参数的平均误差通常低于或接近10%。然而,实验材料研究中常用的密度容许偏差约为2%至3%,因此即使表现最好的模型也没有达到可靠应用所需的精度。
所有模型在部分占位子集上的误差均明显增加,通常是常温平衡体系的两至三倍。即使增加训练数据规模也不能完全解决这一问题。UMA和MACE-OMat使用了远大于传统模型的数据集,模拟完成率接近100%,但密度误差仍分别约为5.3%和6.9%,依然高于实际应用阈值。这说明数据规模扩大能够降低灾难性失败,却不一定按比例提高实验性质预测精度。
密度随时间的演化进一步显示,CHGNet和M3GNet的多数轨迹在整个模拟过程中始终保持较高误差。MACE、SevenNet、MatterSim和Orb则表现出更合理的平衡过程:初始阶段受到热扰动影响,误差短暂升高,随后逐渐收敛到相对稳定的范围。径向分布函数分析得到相似结论,稳定模型能够较好保持短程键合和中程结构相关性,但整体准确度仍有限。
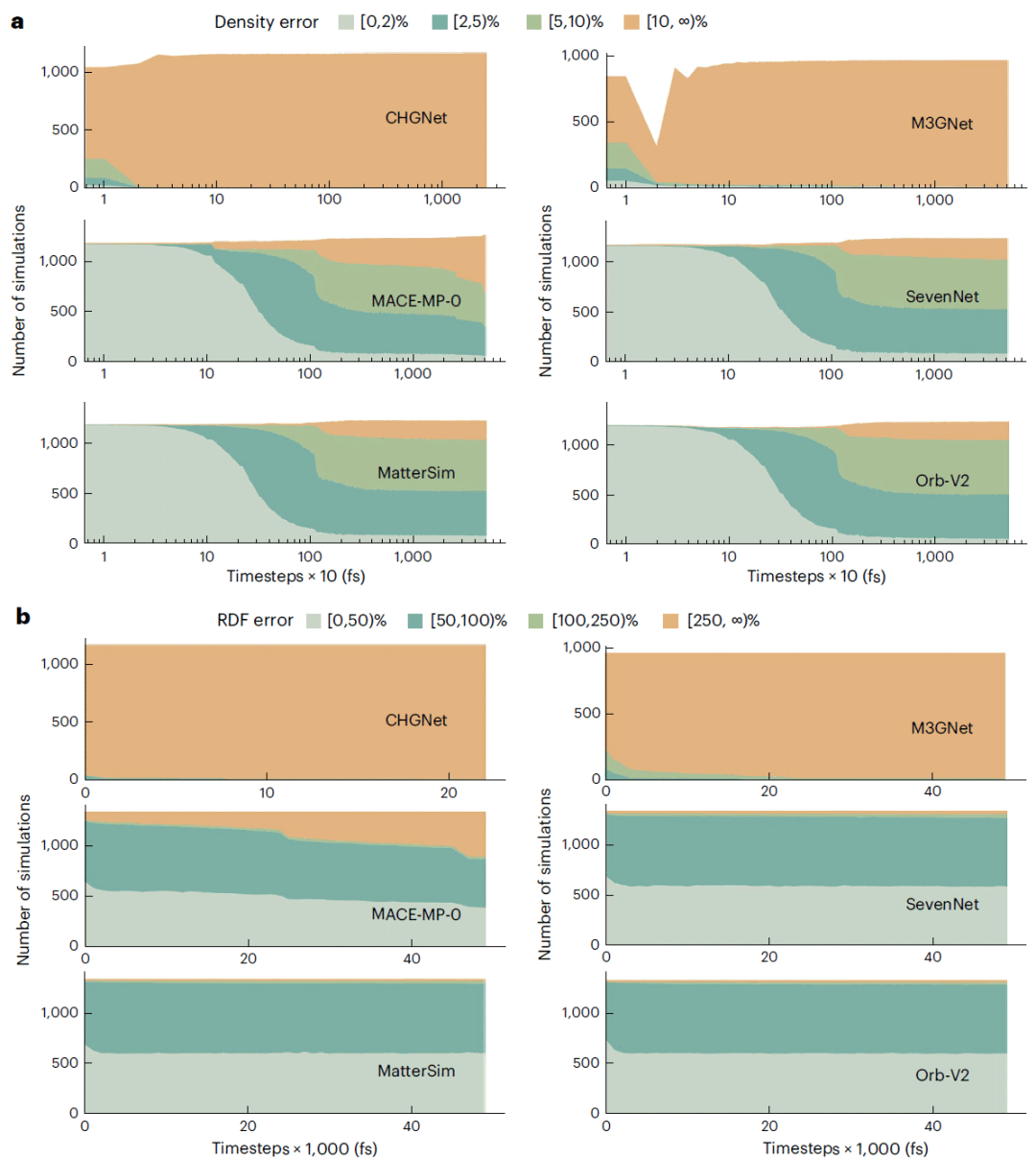
图4:六种模型在分子动力学过程中的密度误差和径向分布函数误差随时间的演化。
弹性性质预测
弹性张量是对模型泛化能力更严格的检验,因为当前模型通常并未直接使用实验弹性数据训练。研究人员对MinX-EM中的100种矿物施加不同方向的小应变,并根据应力响应计算弹性系数。
多数稳定模型对纵向弹性分量的预测相对较好,误差约为20%至25%,但对剪切相关分量的预测明显恶化。即使最佳模型,不同弹性分量的误差仍普遍处于20%至46%。Orb在结构稳定性、密度和键长方面表现突出,却在全部弹性分量上出现严重失效,部分误差接近100%。这表明获得平滑稳定的原子轨迹,并不意味着模型正确学习了势能面对形变的高阶响应。
大规模训练的UMA和MACE-OMat同样没有解决该问题。虽然两者结构优化成功率很高,但杨氏模量和剪切模量的预测误差仍然显著。结果说明,现有训练范式主要优化能量和原子力,尚未充分约束决定弹性性质的能量—应变关系。
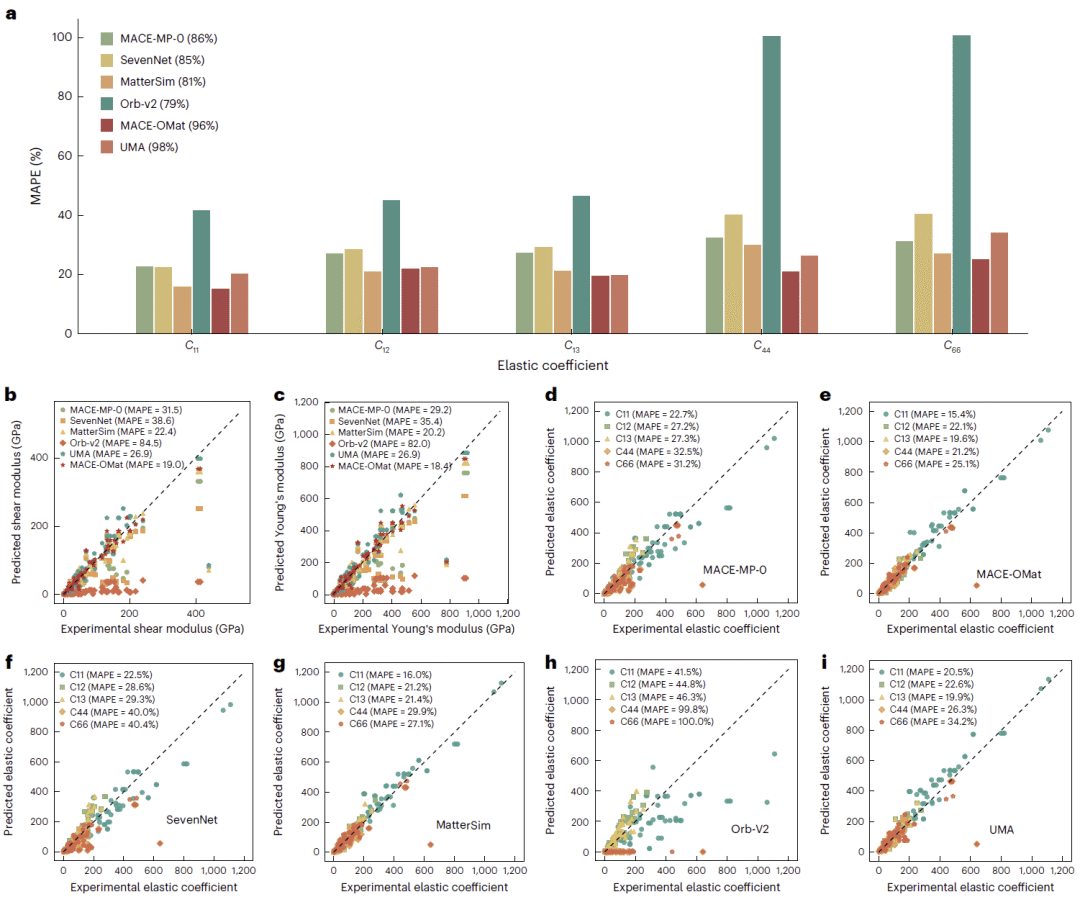
图5:各模型对弹性张量分量、剪切模量和杨氏模量的预测结果及其与实验值的比较。
训练数据偏差与失效机制
元素对分析揭示出明显的氧元素偏差。所有模型对含氧键通常具有较低误差,而对训练数据中较少出现的元素组合预测较差。这与MPtrj及其衍生数据集中氧化物占比较高密切相关。总体而言,某种原子对在训练数据中出现越频繁,其键长误差越低,但高频出现并不足以保证准确性。
部分过渡金属体系还受到参考计算不一致的影响。某些金属仅在与氧或氟结合时使用额外电子校正,导致训练数据中同时存在不完全一致的势能面。模型被迫在这些不同参考之间插值,可能形成非物理的排斥区域,并进一步传播到能量、原子力和材料性质预测中。
相互作用曲线分析表明,CHGNet和M3GNet在短程排斥区域具有高度不平滑的曲率,因此需要极小的积分步长才能维持稳定,这可以解释其高失败率。Orb的相互作用曲线接近经典平滑势,因此能够稳定完成全部轨迹。但平滑性只保证数值稳定,不能保证力随形变的梯度正确,这正是Orb在弹性性质预测中失效的原因。
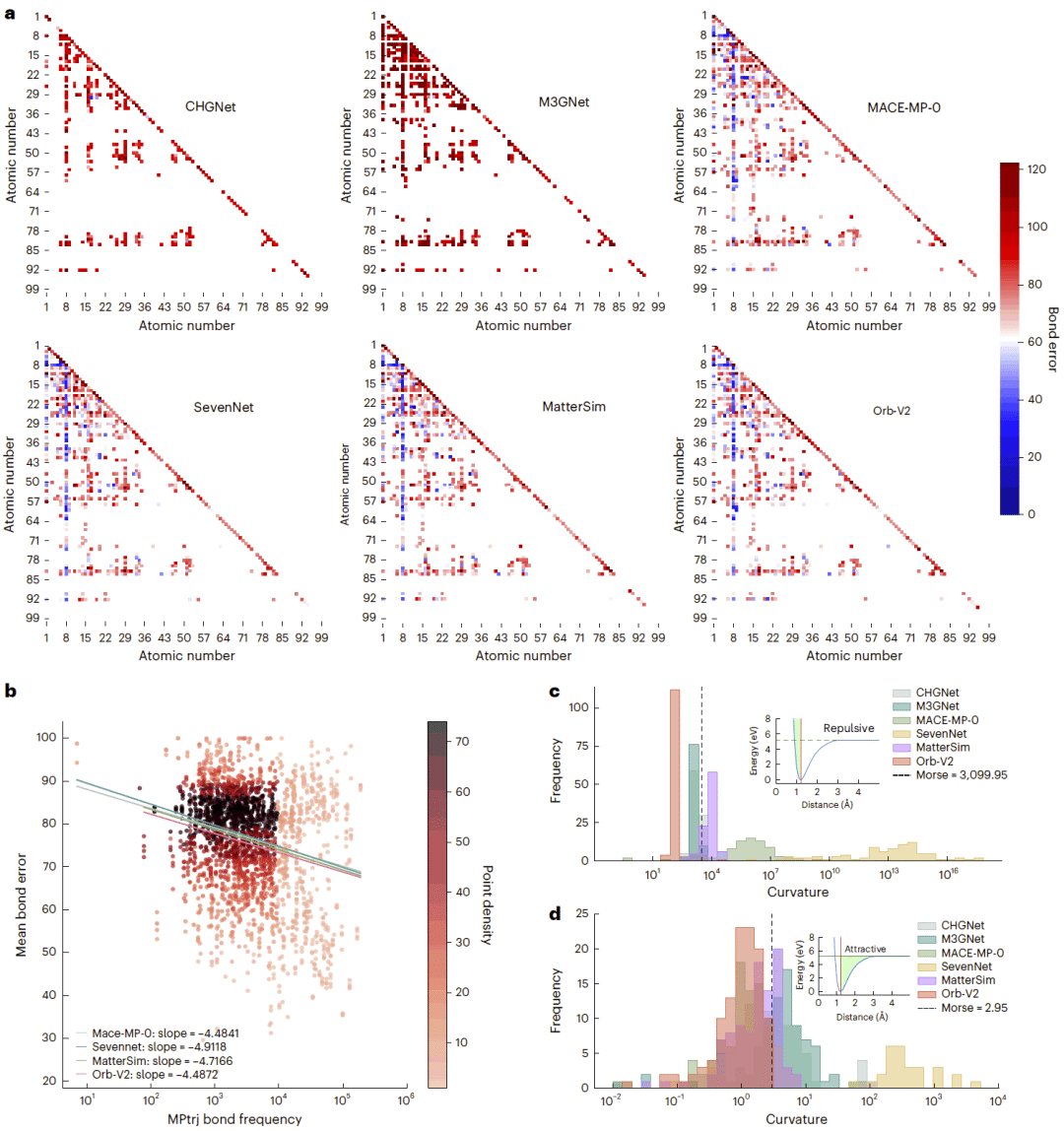
图6:不同模型的元素对键长误差、训练频率与误差关系,以及吸引和排斥区域相互作用曲率分布。
讨论
UniFFBench揭示了传统计算基准成绩与实验应用能力之间的“现实差距”。模型在密度泛函理论能量和原子力任务上的高精度,与其对实验密度、晶格参数、剪切模量和杨氏模量的预测性能相关性很弱。这表明现有评价体系可能高估了模型在真实材料模拟中的可靠性。
模型架构本身并不是唯一瓶颈。不变模型和等变模型都可能获得较高的结构稳定性,说明关键限制更多来自训练目标和训练数据。扩大数据集能够提升模型对未知结构的稳定性,但如果新增数据仍集中于相似化学环境、零温构型和能量—原子力标签,就难以学习有限温度、非谐振动以及机械形变所需的物理规律。
研究人员据此提出,下一代通用机器学习力场应同时学习能量、原子力、应力和实验性质,并通过弹性张量、声子响应和稳定性条件等信息约束势能面的高阶变化。训练集不仅需要覆盖更多元素,还应覆盖更丰富的配位环境、键合方式、温压条件、部分占位和多元素组成。实验基准也应成为模型评价的常规组成部分,而不能只依赖与训练数据来源相同的计算参考。
UniFFBench目前主要面向天然矿物,对合金、玻璃、纳米材料和聚合物的代表性仍有限。其力学评价也以弹性张量为主,未来还可扩展至热膨胀、黏度、热导率和电导率等实验性质。尽管如此,该研究清楚表明,稳定模拟、较低能量误差和较大训练规模均不能单独证明模型具有普适性。真正可靠的通用机器学习力场,必须在复杂实验体系和实际应用指标上接受系统检验。
整理 | DrugOne团队
参考资料
Mannan, S., Bihani, V., Gonzales, C. et al. UniFFBench: evaluating universal machine learning force fields against experimental measurements. Nat Comput Sci (2026).
https://doi.org/10.1038/s43588-026-01019-4
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-16,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号