AlphaFold3 到底有多可靠?五类应用揭示其能力边界、特异性缺口与训练数据依赖

AlphaFold3 到底有多可靠?五类应用揭示其能力边界、特异性缺口与训练数据依赖

DrugIntel
发布于 2026-07-17 20:45:53
发布于 2026-07-17 20:45:53

文献来源
论文题目:Capabilities, specificity gaps and training-data dependence of AlphaFold3 across diverse application areas
作者: Océane Follonier、Yan Liu、Pablo Campomanes、Luc Lafrenaye 等
主要机构: 巴塞尔大学、瑞士生物信息学研究所、洛桑大学、苏黎世联邦理工学院、弗里堡大学等
发表形式: bioRxiv 预印本
发布时间: 2026 年 7 月 13 日
DOI: 10.64898/2026.07.13.738147
论文类型: 跨任务基准评估、应用研究与方法能力边界分析
论文围绕泛素化蛋白、T 细胞受体—表位、抗体—抗原、蛋白质—RNA 和蛋白质—脂质五类任务,对 AlphaFold3 的结构恢复、相互作用特异性、置信度可靠性和训练数据依赖进行了系统评估。
导读
AlphaFold3 将结构预测扩展到蛋白质、核酸、小分子、脂质及翻译后修饰组成的全原子复合物,但能够输出一个几何上合理的复合物,并不等同于能够判断两个分子是否真实相互作用,更不等同于能够预测结合强弱、突变效应或训练集外行为。
这项研究没有提出新的结构预测网络,而是把 AlphaFold3 放入五类真实应用场景中进行压力测试。结果显示,AF3 在泛素化结构建模、部分 TCR 筛选、抗体界面采样和 RNA 结合位点定位中展现出有价值的新能力;但它普遍存在三个问题:置信度难以区分真实配对与诱饵配对,正确结构可能被采样出来却无法被内部排序选中,以及蛋白质—脂质等任务在训练集外明显退化。
这篇论文最值得关注的结论是:AlphaFold3 已经是强大的结构假设生成器,但尚不能被普遍视为相互作用分类器、亲和力预测器或无需校准的虚拟筛选评分函数。
为什么这篇论文值得关注?
AlphaFold2 的成功具有相对清晰的评价标准:给定一条蛋白质序列,模型能否恢复其折叠结构。对于许多稳定的单体蛋白,高 pLDDT 往往确实对应较可靠的局部结构,因此用户可以逐渐建立一套相对成熟的使用规则。
AlphaFold3 面临的情况复杂得多。
它接受的不再只是蛋白质序列,还可以包括 RNA、DNA、小分子、离子、脂质以及共价连接信息。输出对象从一条蛋白质链扩展为包含多类化学实体的全原子复合物。相应地,用户提出的问题也发生了变化:
- • 两个分子是否真正相互作用?
- • 哪一个 RNA、脂质、肽段或抗体才是正确配体?
- • 一个点突变是否会破坏结合?
- • 多次采样得到的结构中,哪一个才是正确界面?
- • 高 ipTM 是否意味着强结合或高特异性?
- • 模型是在迁移一般物理与几何规律,还是在重现训练集中出现过的复合物模式?
这些问题已经超出了传统结构恢复的范围。它们涉及二分类、排序、亲和力、突变效应、分布外泛化和不确定性校准。
因此,这篇工作的核心价值不在于再次报告 AF3 能生成哪些复合物,而在于区分三个经常被混为一谈的能力层级:
能力层级 | 实际问题 | 本文结论 |
|---|---|---|
结构生成 | 能否产生几何合理的复合物? | 多个任务中可以 |
结构选择与相互作用判别 | 能否从候选中选出正确配对和正确界面? | 经常不足 |
分布外决策 | 能否处理训练集中未覆盖的分子、界面和扰动? | 明显依赖任务,部分任务显著退化 |
AF3 在第一层能力上的进步较为明确,但真实科研和药物发现通常需要后两层。
2. 论文评估 AlphaFold3 下游可用性
这不是一篇模型开发论文。作者没有重新训练 AF3,也没有修改其扩散网络或 Pairformer,而是将 AF3 作为一个现成的全原子结构模型,构建了五套不同的应用基准。
整套评估围绕四条主线展开。
2.1 结构是否正确
作者使用 RMSD、界面恢复率、交联位点距离和配体口袋对齐 RMSD,判断预测结构是否接近实验观察。
这一层回答的是:模型能不能把分子放到大致正确的位置。
2.2 模型是否具有特异性
作者不仅输入已知相互作用对,还构建随机 RNA、非 RNA 结合蛋白、非脂质转运蛋白以及不结合的突变体等对照。
这一层回答的是:模型是否会对错误输入同样给出高置信结构。
这是整篇论文最重要的设计。只测量已知复合物的结构恢复,无法区分模型究竟学会了相互作用规律,还是倾向于为任何给定输入构造一个看起来合理的复合物。
2.3 内部置信度能否选出正确答案
AF3 可以通过不同随机种子和扩散采样产生多个结构。作者比较了两种选择方式:
- • 实际选择: 根据 AF3 自身的 ranking score、ipTM 或 ipSAE 选出最高分模型;
- • Oracle 选择: 利用实验真值,在所有样本中挑出真正最准确的模型。
二者之间的差距刻画了一个关键问题:模型可能拥有足够的生成能力,但缺乏可靠的识别和排序能力。
2.4 结果是否依赖训练集重叠
作者采用时间截断、序列同源性过滤和是否存在训练期脂质复合物等方式,构建低同源或分布外子集。
这一层回答的是:准确结果来自可迁移的结构规律,还是来自对相似训练样本的再现。
3. 五类应用是如何测试的?
3.1 泛素化蛋白:通过小分子桥接绕过蛋白—蛋白共价键限制
任务难点
泛素化是在泛素 C 端 Gly76 与靶蛋白赖氨酸侧链之间形成异肽键。AF3 可以指定小分子与蛋白质之间的共价键,却不能直接表达任意蛋白质—蛋白质共价连接。
作者设计了一个巧妙的输入转换:
- 1. 将泛素末端 Gly76 从蛋白链中拆出;
- 2. 把 Gly76 表示为一个小分子配体;
- 3. 用
bondedAtomPairs指定其与剩余泛素链之间的肽键; - 4. 再指定 Gly76 与靶蛋白赖氨酸 ε-氨基之间的异肽键;
- 5. 将重新编码后的体系输入 AF3。
论文图 1A,清楚展示了这一转换:原本无法直接表达的蛋白—蛋白共价键,被拆解为两个由小分子节点连接的共价键。
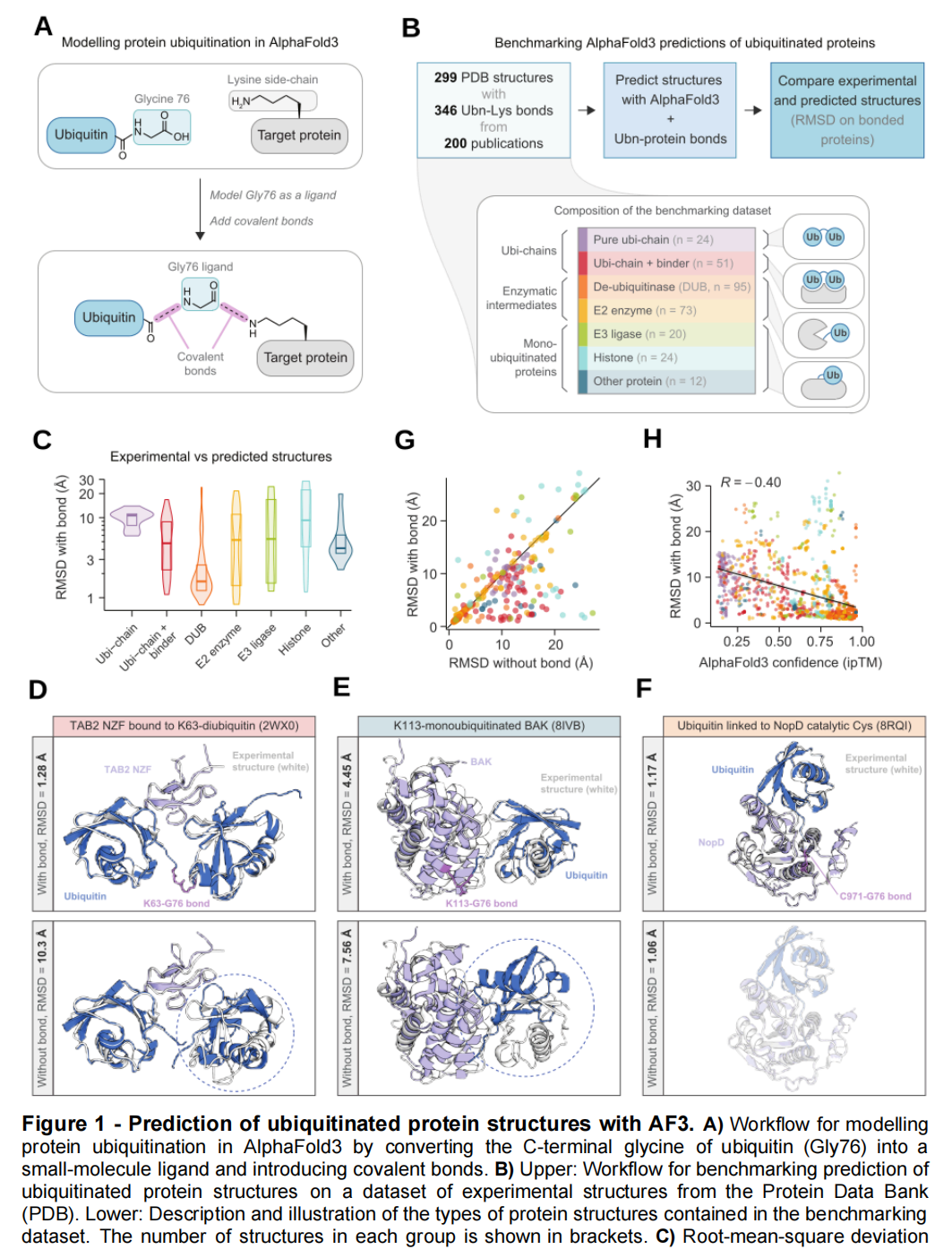

数据与评价
作者从 PDB 整理了:
- • 299 个泛素化或泛素样蛋白结构;
- • 346 条泛素—蛋白共价键;
- • 包括泛素链、去泛素化酶中间体、E2/E3 酶复合物、单泛素化蛋白和组蛋白;
- • 还包含 NEDD8、ISG15 以及半胱氨酸连接等非经典连接形式。
每个体系生成 5 个 AF3 模型,利用 DockQ 确定链对应关系,再计算共价连接蛋白对的 RMSD。
结果
299 个体系中,169 个达到 RMSD 小于 5 Å。
表现最好的类别包括:
- • 去泛素化酶催化中间体:中位 RMSD 1.62 Å;
- • 非组蛋白单泛素化体系:中位 RMSD 4.23 Å;
- • 带非共价结合蛋白的多聚泛素链:中位 RMSD 4.87 Å。
明确输入共价键非常重要。移除连接约束后,泛素经常被放在错误位置或错误方向。图 1D–F 对有无共价键的模型进行了直观比较。
ipTM 在这一任务中具有一定实用价值。ipTM 高于 0.8 的预测约占 44%,其 RMSD 中位数为 1.55 Å;其余预测的中位数为 7.27 Å。
应如何理解?
这是论文中相对积极的一项结果。
首先,AF3 的全原子输入接口可以通过化学重编码扩展到原本不支持的生物共价连接。其次,移除共价约束后性能明显下降,说明成功结果并非全部来自简单记忆。
但其泛化证据仍然有限。经过训练截止日期和 30% 序列一致性过滤后,只得到 11 个较新颖的泛素—蛋白对,样本量不足以判断真正的分布外能力。此外,RMSD 小于 5 Å 对大型蛋白域相对取向而言是较宽松的标准,不能直接保证局部界面、构象变化或功能状态正确。
例如,AF3 未能恢复 NEDD8 连接 Cullin-5 后已知的构象转变。这提示:给定共价拓扑后恢复一个常见复合物构型,与预测修饰诱导的构象重排,仍然是不同难度的问题。
3.2 TCR—表位识别:部分场景可以用于候选优先级排序,但难以预测微小序列扰动
输入与评分方式
每次预测输入完整的:
- • MHC 重链;
- • β2 微球蛋白;
- • 呈递肽;
- • TCR α 链;
- • TCR β 链。
作者预计算 MHC、β2m 和 TCR 链的 MSA,以降低大规模预测成本。每个复合物使用一个随机种子,并保留 AF3 排名最高的模型。
最终评分不是直接采用一个全局 ipTM,而是计算四类链间 ipTM 的平均值:
- • 肽—TCRα;
- • 肽—TCRβ;
- • HLA—TCRα;
- • HLA—TCRβ。
这相当于同时评价 TCR 是否靠近肽段和整个 pMHC 表面。
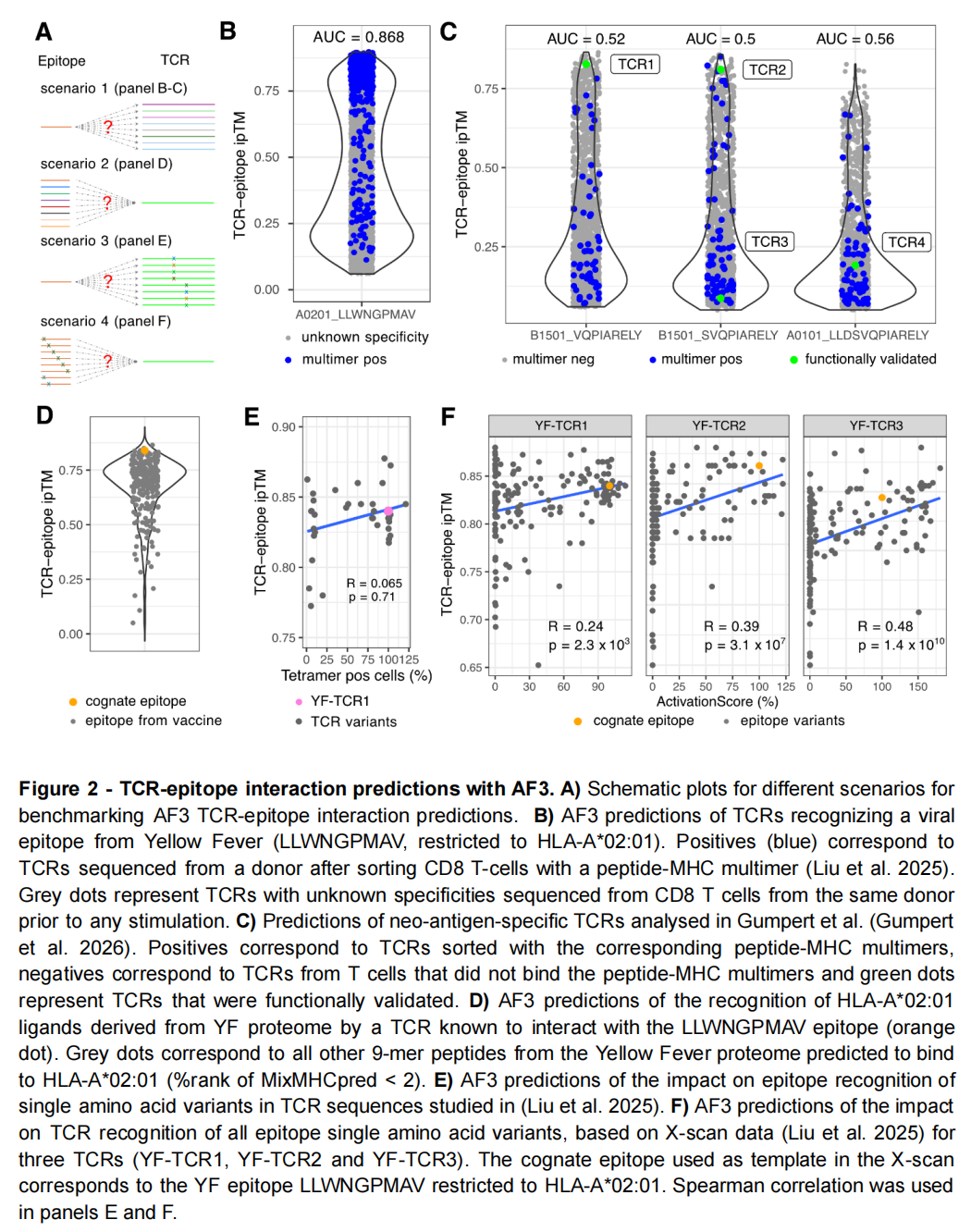
场景一:从 TCR 库中寻找黄热病毒表位特异性 TCR
作者使用同一位接种者的基线 TCR 库作为对照,并以四聚体分选得到的特异性 TCR 作为阳性。
结果的 ROC-AUC 达到 0.88。
这是一个较有说服力的结果,因为阳性与对照来自同一供体和相近实验流程,降低了批次偏差和个体差异造成的伪预测。
场景二:癌症新抗原 TCR
在三个癌症新表位上,整体 AUC 均低于 0.6,说明 AF3 无法稳定地将多聚体阳性 TCR 与阴性 TCR 分开。
但四个经过功能验证的 TCR 中,功能最强的两个分别排在:
- • 1,946 个候选中的第 19 位;
- • 1,673 个候选中的第 22 位。
它们均进入约前 1%–2%,已经接近实际可进行实验验证的规模。
因此,AF3 在这里更像一个高召回的候选缩减工具:它不能保证整体分类性能,但可能把部分强反应 TCR 推到实验可处理的候选范围。
场景三:为已知 TCR 寻找目标表位
作者从黄热病毒多聚蛋白中枚举全部 9-mer 肽段,先用 MixMHCpred 筛选可能结合 HLA-A*02:01 的 251 条肽,再逐一与目标 TCR 共折叠。
真实表位在 251 条候选中排名第 4。
这说明,当上游已经把候选肽空间缩小到合理范围时,AF3 可以提供进一步的结构优先级排序。
场景四:点突变效应
作者分别测试:
- • TCR CDR3 位点的单氨基酸替换;
- • 表位肽每个位置的系统性氨基酸扫描。
AF3 预测分数与实验激活或结合结果仅呈弱相关。许多已经完全破坏 TCR 识别的变体仍然获得较高 ipTM。
论文图 2(PDF 第 8 页)非常直观:黄热病毒任务可以得到较好的总体区分,而新抗原任务的阳性和阴性分布高度重叠;突变扫描中,回归趋势远弱于散点噪声。
核心判断
AF3 能够捕捉某些 TCR—pMHC 的整体几何兼容性,但对决定免疫识别的细粒度能量差异不够敏感。
点突变可能只改变少数氢键、疏水接触、去溶剂化代价或构象熵,整体复合物仍然能够被模型构造成结构合理的状态。ipTM 主要反映模型对相对结构的信心,并不是自由能差或功能激活概率。
3.3 抗体—抗原:生成能力强于排序能力
为什么难?
抗体结合界面主要由 CDR 环构成,尤其是 CDR-H3。它们缺少一般蛋白复合物中常见的共进化信号,柔性大,并且多个表面区域都可能形成几何上合理的接触。
作者选择 SARS-CoV-2 RBD 抗体数据,覆盖 D614G 与 Omicron BA.1 两种病毒株。由于 Omicron BA.1 出现于 AF3 训练截止日期附近,相关复合物可用于一定程度的时间外测试。
大规模采样方案
作者先通过 100 个随机种子测试采样收敛,发现约 50 个种子后结果趋于稳定。
随后针对每个抗体—RBD 体系运行:
- • 50 个随机种子;
- • 每个种子生成 5 个扩散样本;
- • 总计每个靶标 250 个 AF3 结构。
AF2-Multimer 则使用 1 个种子、5 个模型。
作者比较:
- • AF3 自身评分选出的 top 模型;
- • 250 个模型中按真实界面挑出的 best 模型。
最关键的结果:正确答案经常存在,但 AF3 选不出来
在 50 个种子生成的模型中,正确抗原表位几乎总能在某个样本中出现。但是,AF3 的排名分数通常没有把该模型排到首位。
使用基于 PAE 的 ipSAE 也没有显著解决这一问题。
论文图 3中,上半部分显示按模型自身分数选出的成功率,下半部分显示利用真值进行 oracle 选择的成功率。两者之间的明显差距揭示了一个重要事实:
AF3 的构象搜索空间中可能已经包含正确界面,主要瓶颈却转移到了结构选择和置信度校准。
AF3 在 16 个表位类别中的 10 个上优于 AF2,并且对传统表位可预测性指标 Discotope 的依赖较小。这说明它确实扩展了抗体界面的采样能力。
但 AF3 的界面恢复率与抗体亲和力没有明显相关性,对破坏结合的 RBD 点突变也不敏感。
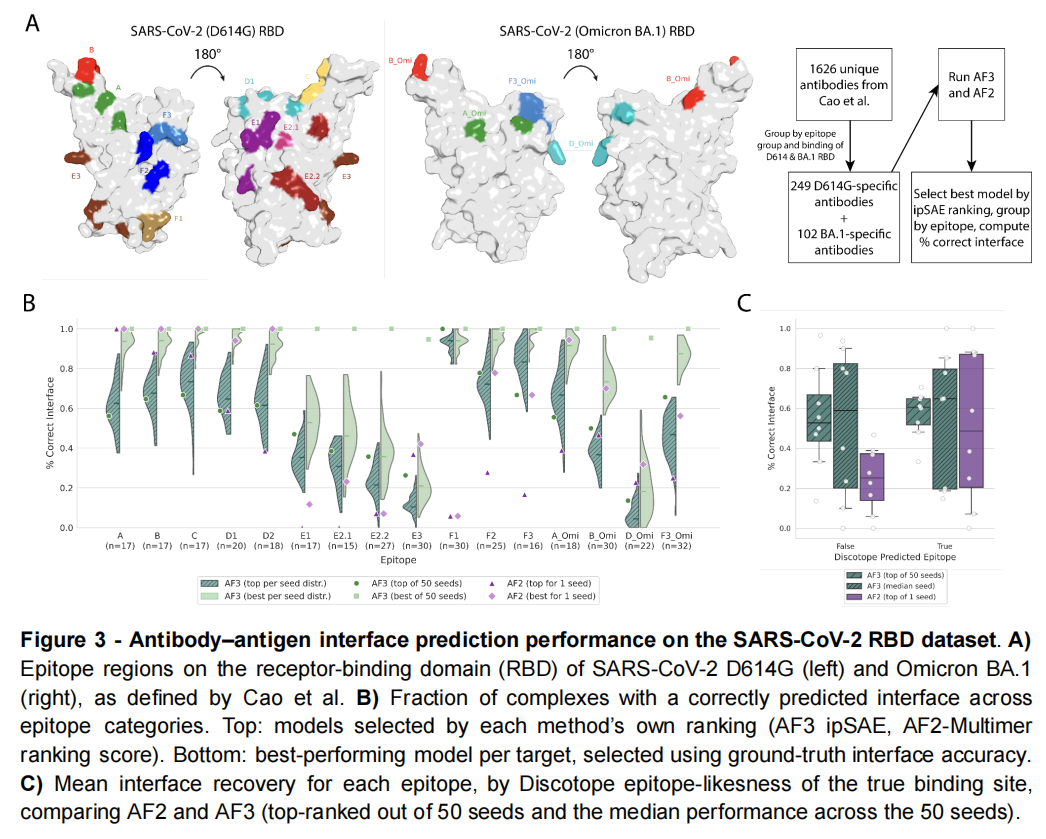
为什么会这样?
共折叠模型通常在已知发生相互作用的复合物上训练。模型学习的问题近似于:
已知这些分子形成复合物,它们可能采用什么结构?
而虚拟筛选需要回答:
这些分子究竟会不会结合?若会结合,结合有多强?
两个任务的条件分布不同。缺少不结合复合物和困难负样本训练时,模型容易为错误配对也构建出一个自洽界面。
因此,结构置信度不应被直接解释为抗体结合概率,更不能替代亲和力或逃逸突变模型。
3.4 蛋白质—RNA:可以找到 RNA 结合区域,却难以识别正确 RNA
这是整篇论文中最能说明结构定位与分子特异性分离的实验。
数据如何构建?
作者整合了三类证据:
- 1. POSTAR 中的 CLIP-seq 数据,用于确定 RNA 结合蛋白的 RNA 靶标;
- 2. RNPxl 和 XRNAX 的交联质谱数据,用于确定蛋白侧接触残基;
- 3. CPTAC 的 1,072 个肿瘤样本,通过拷贝数、转录组和蛋白组的联合关联获得癌症相关 RBP—RNA 对。
CPTAC 部分要求同一 RBP—RNA 对在三个组学层面均达到校正后 (p<0.01),且效应方向一致,最终得到 343 个稳健关联。
作者构建三组预测:
分组 | 组成 | 数量 |
|---|---|---|
真实配对 | 实验或多组学支持的 RBP—RNA | 369 |
RBP + 随机 RNA | RNA 结合蛋白与错误 RNA 配对 | 510 |
非 RBP 对照 | 无 RNA 结合注释的蛋白与 RNA | 230 |
所有 RNA 长度统一为 25 个核苷酸,避免模型仅依靠长度差异进行区分。
AF3 学到了什么?
三组平均 ipTM 分别为:
- • 真实 RBP—RNA:0.437;
- • RBP + 随机 RNA:0.398;
- • 非 RBP + RNA:0.305。
AF3 能够区分 RNA 结合蛋白与非 RNA 结合蛋白,但真实 RNA 与随机 RNA 之间的差距很小,而且主要受到两组蛋白组成差异的影响。
这表明 AF3 更容易识别:
这个蛋白是否具有适合容纳 RNA 的折叠和表面。
它较难回答:
这条具体 RNA 序列是否是该蛋白的真实靶标。
换句话说,模型获得了较强的结合位点先验,但没有形成可靠的序列配对特异性。
训练集外是否仍然有效?
作者使用 30% 全局序列一致性构建低同源子集。令人意外的是,移除同源模板后 ipTM 没有下降,部分对照组甚至略有升高。
这支持一个相对积极的解释:RNA 结合区域的定位不完全依赖模板复制,模型可能学习到了可迁移的 RNA 结合折叠和表面特征。
界面位置预测相当准确
针对 655 个具有 134 个质谱交联位点的预测,RNA 与实验交联残基之间的最短距离达到:
- • 3 Å 内:77%;
- • 5 Å 内:88%;
- • 10 Å 内:91%。
与随机残基基线相比,中位距离改善约 2.63 Å;低同源子集中改善约 2.81 Å。
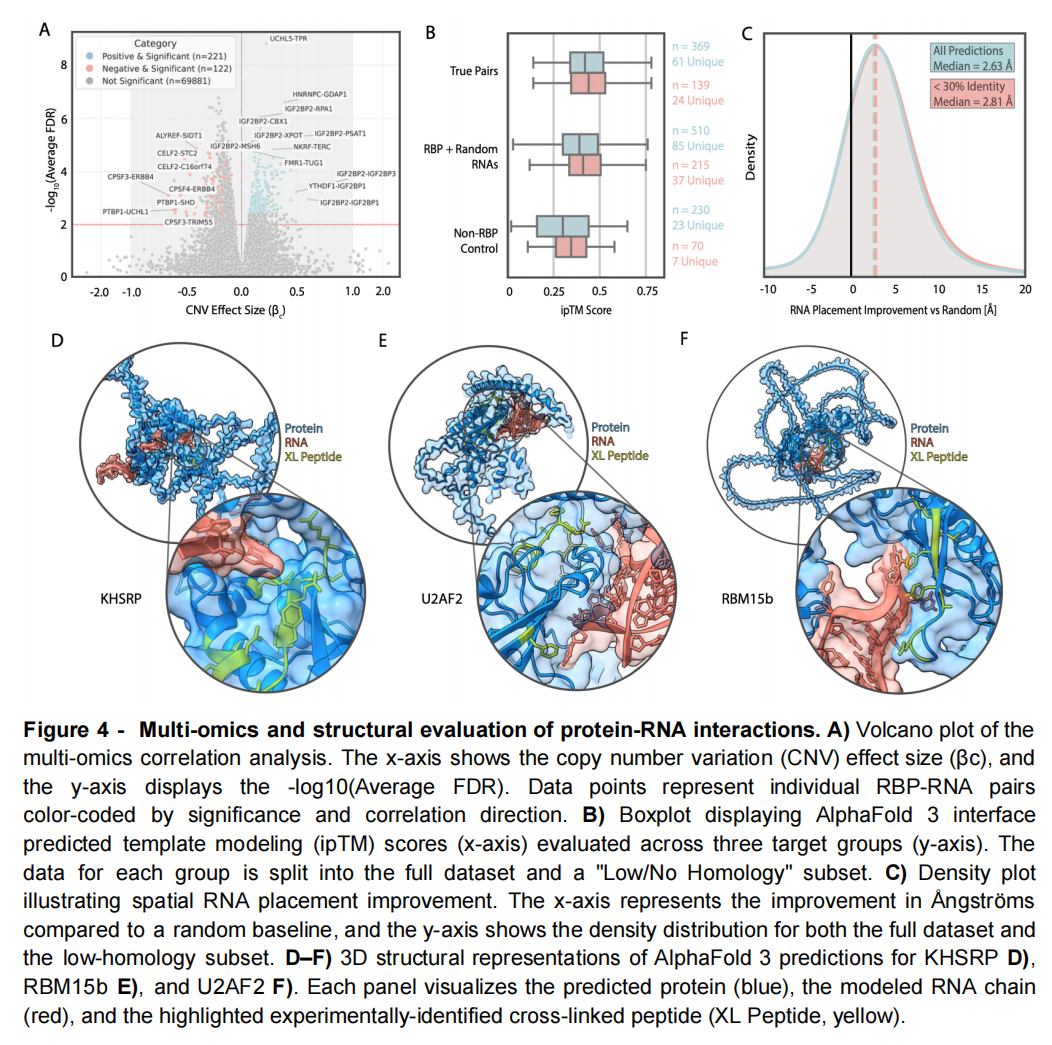
不过,交联位点接近只能证明界面区域合理,不能证明 RNA 的方向、碱基配对、局部原子接触和序列选择性正确。
最准确的概括
AF3 可以较好地回答 RNA 大致在哪里结合,却不一定知道应该是哪条 RNA。
3.5 蛋白质—脂质:训练集内几乎精确复现,训练集外筛选性能显著下降
作者为脂质任务设计了多层次数据集。
DB1:已知脂质转运蛋白与共晶脂质
包含 12 个不同折叠类型的脂质转运蛋白,并为每个蛋白输入:
- • 其共晶脂质;
- • 其他脂质家族成员;
- • 正构烷烃对照。
模型仅接收蛋白质序列和脂质 SMILES,不提供模板。每个配对采用一个随机种子生成 5 个扩散样本,再由 AF3 ranking score 选择首位结构。
对于训练集中存在相关结构的共晶配对,口袋对齐后的脂质平均原子 RMSD 仅为 0.08 Å,接近精确重建。
共晶脂质按照 ipTM 排名:
- • 12 个蛋白中有 6 个排第 1;
- • 11 个进入前 3。
但许多替代脂质同样获得 ipTM 大于 0.8。由于部分脂质转运蛋白可能本身具有多配体能力,单靠这一数据集无法区分模型假阳性与真实结合广谱性。
DB2 和 DB3:大空腔蛋白负对照
作者选取 9 个具有较大内部空腔、但不承担脂质转运功能的蛋白,与多类脂质组成 50 个负样本。
约 7% 的负样本仍获得 ipTM 大于 0.8。
将 12 个共晶阳性与 50 个负样本合并后,AUPRC 为 0.701。这说明 AF3 在简化场景中具有中等偏好的区分能力,但无法完全排除仅凭大空腔和疏水环境形成的伪结合模式。
DB4 与 DB5:大规模实验筛选
作者进一步使用 1,394 个蛋白质—脂质配对:
- • 125 个实验阳性;
- • 1,269 个阴性或未检测到结合的配对。
根据 AF3 训练期是否出现相关脂质结合结构,划分为:
- • DB4: 615 对,含训练重叠蛋白;
- • DB5: 779 对,无脂质复合物训练重叠。
结果如下:
数据集 | AUPRC | NEF@1% | NEF@5% | NEF@10% |
|---|---|---|---|---|
DB4,存在训练重叠 | 0.309 | 0.86 | 0.35 | 0.34 |
DB5,分布外 | 0.189 | 0.50 | 0.15 | 0.18 |
训练重叠组在最前 1% 候选中具有较强富集能力,但整体精度仍然有限。进入分布外数据后,AUPRC 和早期富集均明显下降。
论文图 5把实验阳性点叠加在所有 ipTM 小提琴分布上。DB1 中共晶脂质大多位于高分区域;到了 DB4 和 DB5,阳性与阴性出现大范围重叠,且不少真实相互作用低于常用的 0.8 阈值。
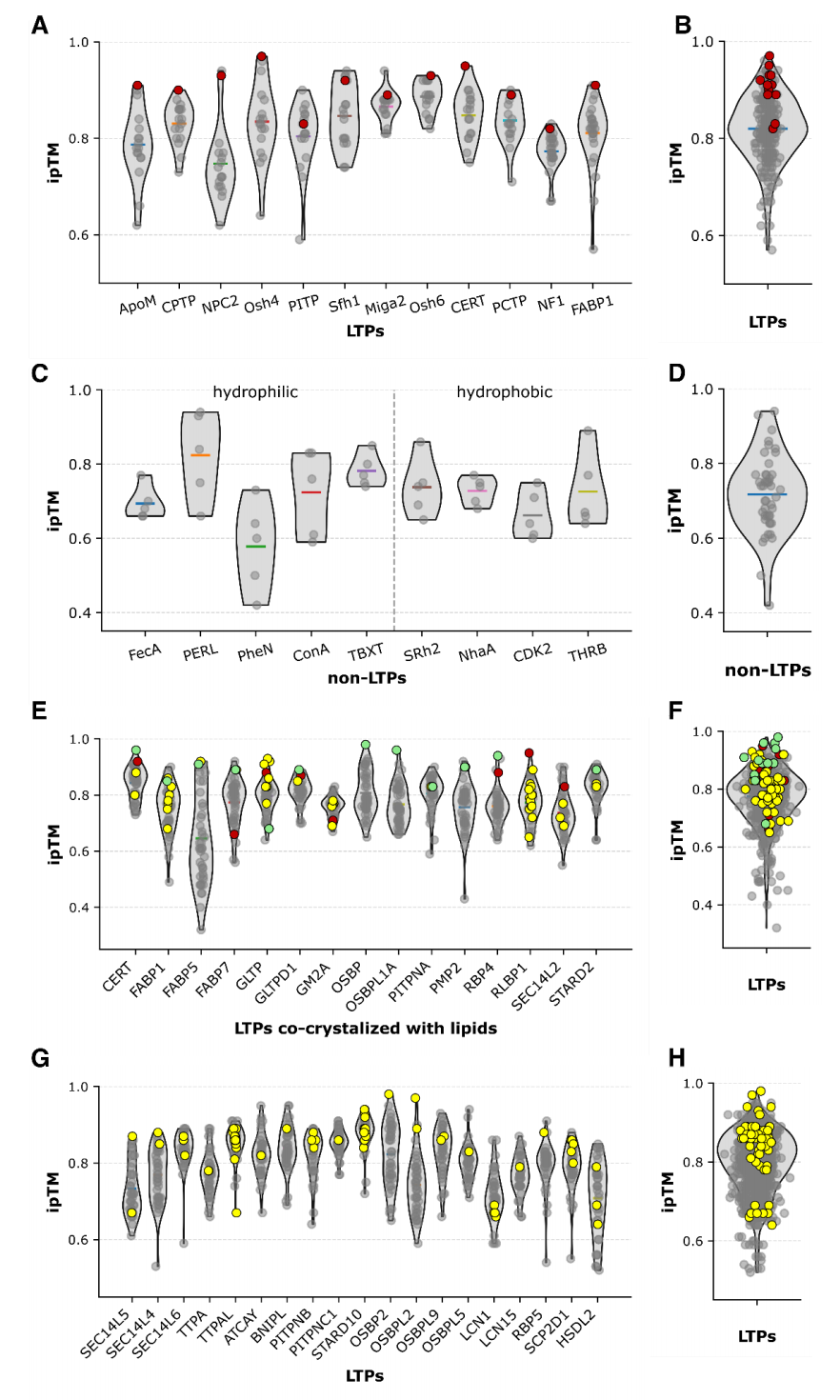
这说明什么?
AF3 能够非常准确地重建已知蛋白质—脂质结构模式,也能在训练分布附近富集部分阳性,但这种能力尚不足以支持无校准的大规模脂质筛选。
特别需要注意,**接近完美的已知结构 RMSD 与较低的分布外 AUPRC 可以同时出现。**前者证明模型善于重现熟悉结构,后者才更接近实际发现任务。
4. 五类任务的结果放在一起,真正的规律是什么?
应用 | AF3 的主要优势 | 主要缺口 | 更合适的定位 |
|---|---|---|---|
泛素化蛋白 | 显式共价约束后可恢复大量结构,ipTM 有一定筛选价值 | 修饰诱导构象变化和真正 OOD 证据不足 | 结构假设生成 |
TCR—表位 | 部分任务可将真实 TCR 或表位排入实验可测范围 | 跨表位不稳定,点突变效应弱 | 候选优先级排序 |
抗体—抗原 | 多次采样经常覆盖正确界面 | 内部排名选不出正确样本,不反映亲和力 | 界面采样器 |
蛋白质—RNA | RNA 结合区域定位较准确 | 无法可靠识别正确 RNA 序列 | 结合位点预测 |
蛋白质—脂质 | 训练分布内结构恢复极强,具有一定早期富集 | 假阳性多,OOD 性能明显下降 | 初筛与假设生成 |
这五类任务共同指向同一个问题:
AlphaFold3 更擅长回答结构能否被构造,而不总能回答相互作用是否真实发生
当输入一个蛋白和一个候选分子时,AF3 倾向于寻找一个在训练分布和几何先验下较为自洽的复合物构型。只要蛋白表面存在合适口袋、疏水空腔或核酸结合折叠,错误配体也可能得到合理结构和较高置信度。
这一现象在蛋白质—RNA 和蛋白质—脂质任务中尤其明显:
- • RNA 结合蛋白与随机 RNA 仍然获得较高 ipTM;
- • 非脂质转运蛋白的大空腔仍然可能得到高置信脂质姿势。
模型的结构先验可能是正确的,但它缺少足够的证据判断该分子是否是生物学上的真实配对。
5. 为什么 ipTM 不能被直接当作结合分数?
ipTM 最初用于估计复合物链间相对构型的可靠性。它衡量的是模型对界面几何的结构信心,通常受到预测对齐误差分布的影响。
它并不直接计算: 结合自由能;解离常数;去溶剂化代价;构象熵;竞争配体;细胞内浓度;动力学过程;非结合状态的稳定性。
一个错误配对只要能够形成类似训练集中复合物的界面,也可能得到较高 ipTM。
本文提供了三个直接证据:
- 1. 抗体界面恢复与实验亲和力不相关;
- 2. 破坏 TCR 或抗体结合的单点突变仍可能保持高分;
- 3. RNA 结合蛋白与随机 RNA 的分数接近真实配对。
因此,在小分子对接、抗体筛选、TCR 发现和 RNA 靶标筛选中,ipTM 更适合被解释为:
模型认为该输入可以形成一个稳定、可识别的结构模式。
它不应被自动解释为:
该输入在真实生物环境中一定结合,且分数越高结合越强。
6. 这篇文章的技术启发
6.1 结构恢复、相互作用识别和亲和力预测必须分别评估
在已知阳性复合物上计算 RMSD,只评价模型能否恢复条件分布中的结构。
要评价虚拟筛选能力,至少需要同时加入: 实验阴性;随机配对;具有相似口袋或表面的困难负样本;活性悬崖和单点破坏突变;时间外与结构外测试集。
一个模型可以在 RMSD 上表现优秀,却在 AUPRC 和早期富集上表现一般。脂质任务正是典型案例。
6.2 生成正确答案不代表能够识别正确答案
抗体任务显示,增加随机种子后,正确界面经常已经出现在 250 个候选结构中,但模型自身分数无法把它选出来。
未来模型必须同时改进两个相互独立的模块:
- • 采样器: 是否覆盖正确构象;
- • 选择器: 是否能够从样本中识别正确构象。
只报告 oracle best-of-N 会高估实际可用性,只报告 top-1 又可能低估模型内部已经学习到的结构能力。二者应当同时给出。
6.3 缺少负样本是共折叠模型的根本限制之一
如果训练数据主要由已知复合物构成,模型得到的监督信号是如何安排已经相互作用的实体。
这类数据无法充分教会模型拒绝:错误 RNA;非结合抗体;错误脂质;不相容小分子;破坏结合的突变体。
未来的全原子结构基础模型需要引入经过实验约束的非相互作用数据、相对亲和力数据和竞争关系,而不能只依赖 PDB 阳性结构。
6.4 训练数据重叠不能只用序列一致性衡量
30% 序列一致性过滤能够排除明显同源模板,但仍可能保留:
- • 相同折叠;
- • 相似界面几何;
- • 相同抗原表面;
- • 相似脂质骨架;
- • 近似口袋;
- • 同一蛋白家族中的保守结合模式。
真正严格的分布外评价应同时控制序列、折叠、界面、配体化学结构和发布时间。
6.5 点突变是检验模型是否理解相互作用能量的高价值测试
大幅改变全局结构的问题较容易被结构模型识别。真正困难的是:全局构型几乎不变,但结合能发生显著变化。
TCR 和抗体突变结果说明,AF3 尚未稳定掌握这类局部能量差异。点突变扫描、活性悬崖和同系物竞争应成为未来结构基础模型的标准评测,而非附加实验。
局限性
1. 五类基准的实验真值并不等价
泛素化和部分脂质任务具有直接实验结构;RNA 任务主要依赖 CLIP-seq、质谱交联和多组学相关;大规模脂质任务中的阴性可能只是实验条件下未检测到结合。
因此,不同任务的成功率和分数不能直接横向比较。
尤其是蛋白质—RNA多组学关联并不等同于直接物理结合,脂质未检出也不一定是真正阴性。标签噪声可能同时降低模型表观性能和夸大假阳性。
2. 训练数据依赖的定义不完全统一
泛素化使用时间截断和序列同源性,RNA 使用 30% 全局序列一致性,脂质则根据是否存在训练期脂质结合结构划分。
这些规则回答的不是完全相同的问题。论文已经发现训练依赖,但尚未建立一个统一、可量化的记忆程度指标。
3. 泛素化的真正新颖样本太少
经过严格过滤后只有 11 个结构。当前结果证明工作流可行,但无法充分证明模型能处理全新的泛素连接类型、靶蛋白折叠和修饰诱导构象变化。
4. 抗体任务的计算预算与基线不完全对称
AF3 每个体系使用 50 个种子、250 个结构,而 AF2-Multimer 只使用一个种子和 5 个模型。大量采样本身会提高覆盖正确界面的概率。
这不否定 AF3 的能力,但意味着 best-of-250 与 AF2 top-of-5 不能被简单解释为架构层面的等预算比较。
此外,论文对正确界面的定义为预测接触残基落在真实界面残基前后两个序列位置内,标准相对宽松,不能替代严格 DockQ、iRMSD 或完整接触恢复率。
5. RNA 随机对照仍可进一步加强
随机 RNA 被统一为 25 nt,有助于控制长度,但仍需进一步匹配:核苷酸组成;二级结构;亚细胞定位;表达丰度;是否含已知结合基序。
更困难的结构匹配诱饵可能使特异性缺口更加清晰,也可能揭示模型究竟依赖哪些 RNA 特征。
6. 只评估了 AF3
论文没有系统比较 Chai-1、Boltz、Protenix、OpenFold3 等全原子共折叠模型。因此,当前结果主要说明 AF3 的行为,尚不能自动推广到所有后续模型。
不过,抗体点突变等问题已在相关共折叠模型中出现,说明部分缺陷可能来自共同的训练目标和数据结构,而不只是单个实现。
7. 缺少由模型直接驱动的前瞻性验证
论文使用了大量已有实验数据,但没有选择 AF3 新提出的高置信相互作用进行专门实验验证。
真正的应用闭环还需要回答:
- • 高分但未有实验注释的配对中有多少是真阳性?
- • 低分已知阳性是否对应特殊构象或实验条件?
- • 模型筛出的 TCR、抗体或脂质配对能否提高真实实验命中率?
对 AI 制药和小分子设计的未来意义
这篇研究虽然没有直接评估药物小分子,却对 AF3 类模型在小分子生成、对接和虚拟筛选中的使用具有直接警示。
1. AF3 可以作为姿势生成器,但不能默认作为结合判别器
在蛋白质—配体任务中,模型可能为一个不结合的分子构造出几何上自洽的姿势。由此得到的高 ipTM、低 PAE 或高模型置信度,只能支持结构可行性,不能单独证明活性。
更稳妥的流程应当是:
AF3 共折叠或生成姿势 → 化学与价态检查 → 物理能量精修 → 与困难诱饵比较 → 亲和力或自由能计算 → 实验验证。
2. 用 AF3 自洽性筛选生成分子时,必须警惕确认偏差
一种常见设想是:先由生成模型设计分子,再用 AF3 类模型重预测复合物,并用 ipTM、PAE 或结构一致性筛选。
这一流程可以排除部分几何不稳定设计,但生成器和验证器可能共享类似训练数据和结构先验。二者达成一致,不一定代表分子真实结合,也可能只是共同偏好某种熟悉的口袋—配体模式。
因此,自洽性应被视为一级过滤,而非最终证据。
3. 未来模型应从结构条件生成转向相互作用对比学习
下一代全原子基础模型需要同时学习:正确复合物结构;错误配对不能稳定结合的证据;同一配体对不同蛋白的选择性;同一蛋白对近似配体的活性差异;点突变造成的相对自由能变化;实验条件和分子状态带来的不确定性。
训练目标也需要由单一结构去噪扩展为结构生成、相互作用分类、相对排序、能量学习与置信度校准的联合目标。
4. 未来基准应以决策价值而非单一 RMSD 为中心
对于药物发现,一个更完整的评价框架应包括:
评价维度 | 推荐问题 |
|---|---|
结构恢复 | 已知活性配体的结合姿势是否正确? |
特异性 | 非活性同系物是否被排除? |
排序 | 活性系列中的相对顺序是否正确? |
泛化 | 新骨架、新靶标和新口袋上是否保持性能? |
局部敏感性 | 活性悬崖和单原子改动能否识别? |
置信度 | 分数是否与错误概率校准? |
前瞻验证 | 是否提高真实合成与实验命中率? |
这也是 AF3 从结构预测工具走向可靠分子发现基础设施必须跨越的门槛。
总结
这篇论文没有否定 AlphaFold3 的价值。相反,它展示了 AF3 若干过去难以实现的新能力:
- • 通过共价重编码预测泛素化蛋白结构;
- • 从大规模 TCR 库中富集部分真实表位特异性 TCR;
- • 在多次采样中覆盖正确抗体表位;
- • 在低同源场景下定位 RNA 结合区域;
- • 高精度重建熟悉的蛋白质—脂质复合物。
但作者同样清楚地表明,这些能力尚未自动转化为可靠的相互作用决策:
- • 高置信度不一定代表正确分子配对;
- • 正确结构被采样出来后,内部评分仍可能选错;
- • 结构置信度不能替代亲和力;
- • 点突变效应仍然难以预测;
- • 训练集外性能可能显著下降。
因此,现阶段对 AF3 最准确的定位是:
它是一种强大的全原子结构假设生成器和候选优先级工具,适合嵌入包含负对照、物理计算、任务特异性校准和实验验证的完整工作流。
它距离普适的相互作用判别器、亲和力评分函数和无需校准的虚拟筛选平台,仍有实质性距离。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-17,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号