Nat. Comput. Sci. | 理解语言模型规模扩展对蛋白质适应度预测的影响

Nat. Comput. Sci. | 理解语言模型规模扩展对蛋白质适应度预测的影响

DrugAI
发布于 2026-07-14 14:09:49
发布于 2026-07-14 14:09:49
DRUGONE
蛋白质语言模型以及融合蛋白质结构或同源序列信息的模型,通常通过估计蛋白质序列出现的可能性来刻画蛋白质适应度景观,并据此预测突变效应或辅助蛋白质设计。按照深度学习领域的普遍经验,扩大模型参数量和训练数据规模通常能够提升模型能力。然而,在蛋白质适应度预测任务中,这一规律并不总是成立。多个系列模型均表现出类似现象:中等规模模型往往优于更大的模型,模型继续扩展后,预测性能反而下降。
研究人员系统分析了模型规模、训练数据、随机训练因素以及序列可能性与适应度预测之间的关系。结果表明,模型给出的野生型序列可能性不仅由真实蛋白质适应度决定,还会受到训练集中同源序列数量、最近同源序列相似度、模型参数量以及随机初始化和数据顺序等因素影响。当模型对野生型序列的置信度过低时,各种突变的预测分数都接近中性;当置信度过高时,几乎所有突变都会被预测为强烈有害。这两种极端情况都无法真实反映蛋白质适应度景观。
进一步分析发现,多数蛋白质在中等序列可能性水平下获得最佳预测性能。较大模型通常会赋予野生型序列更高可能性,可能越过最佳区间,导致性能下降。真正决定预测质量的并不是模型大小本身,而是模型预测的残基可能性是否与同源序列中的进化保守模式相一致。该研究解释了蛋白质模型在适应度预测任务中的非单调规模扩展规律,并为模型选择和下一代预测器设计提供了实用建议。

蛋白质适应度景观描述不同氨基酸突变如何影响蛋白质的功能、稳定性、表达量、活性和相互作用。准确刻画这一景观对于理解疾病相关变异、监测病毒演化以及指导蛋白质工程具有重要意义。深度突变扫描实验可以一次性测量大量突变,但实验成本高、周期长,并且通常只能测量容易建立实验读出的功能。因此,研究人员一直希望通过计算模型实现大规模、零样本的突变效应预测。
近年来,蛋白质语言模型通过在海量蛋白质序列上进行自监督学习,逐渐成为重要方法。这类模型通常学习一个序列分布,并估计野生型序列和突变序列的可能性。突变序列相对于野生型序列的对数可能性变化,可以作为突变适应度的预测分数。除纯序列语言模型外,还出现了利用多序列比对、三维结构或序列—结构联合信息的模型。
这些方法大致可以分为两类。第一类是通用模型,它们在来自多个蛋白质家族的数百万甚至数十亿条序列上训练,并希望学习可迁移的普遍规律。第二类是家族特异模型,它们针对某一个蛋白质家族的多序列比对单独训练,直接学习该家族的进化保守模式。通用模型更易使用,但其预测可能受到训练数据组成和模型训练过程的复杂影响;家族特异模型更贴近具体进化背景,却需要为每个蛋白质单独准备同源序列和训练模型。
在自然语言和通用人工智能领域,增大模型规模通常会降低训练损失并提升下游性能。蛋白质模型在序列补全、结构预测和序列生成方面也表现出明显的规模效应。然而,在零样本适应度预测中,多个模型系列都显示,中型模型优于最大模型。这一反常现象提示,训练损失更低或序列概率更高,并不必然意味着更准确的突变效应预测。
方法
研究人员以ProteinGYM中的深度突变扫描数据为主要评估基准,筛选出154项实验,共覆盖约48.7万个单残基替换。这些实验涉及活性、结合、稳定性、个体适应度和表达等不同功能类别。研究重点比较了六种不同规模的ESM2模型,并进一步分析ESM1v、ESMC、CARP、ProGen3、RITA、ESM3、SaProt、ProSST、ESM-IF1、MSA-Transformer、EVE和位点独立模型等多种架构。
对于掩码语言模型,研究人员主要采用掩码边际方法,在突变位点遮盖野生型氨基酸后,比较模型对突变氨基酸与野生型氨基酸的预测可能性。对于自回归模型和逆折叠模型,则使用完整序列可能性进行比较。研究人员还分析了训练集中同源序列的数量和相似度、蛋白质结构中的相对溶剂可及性、每个位点的有害突变数量,以及模型预测与家族特异进化模式之间的一致性。为避免复杂突变带来的方法差异,研究仅关注单残基替换。
结果
通用模型的序列可能性受到适应度之外因素影响
研究人员首先考察模型预测的野生型序列可能性是否真正只反映蛋白质适应度。结果显示,训练集中同源序列越多,模型通常会赋予该蛋白质更高的序列可能性;当训练集中存在高度相似的同源序列时,也更容易获得较高预测值。然而,这种关系并不稳定,有些具有大量同源序列的蛋白质仍然被赋予较低可能性,而部分高可能性蛋白质也未必拥有大量近缘同源序列。
模型规模同样显著影响预测。随着ESM2参数量增加,野生型序列的平均可能性持续升高,但不同蛋白质受到的影响幅度差异很大。有些蛋白质在模型扩展后变化很小,另一些蛋白质的预测则大幅提高,这种变化无法仅由同源序列数量或最近同源序列相似度解释。
研究人员进一步比较了结构相同、训练数据相同但随机种子不同的五个ESM1v模型,发现约10%的蛋白质在不同随机训练中出现明显的序列可能性差异。这说明参数初始化、数据打乱和掩码采样等随机因素也会影响最终预测。综合来看,通用模型的序列可能性是一种复杂输出,不能简单等同于真实适应度。
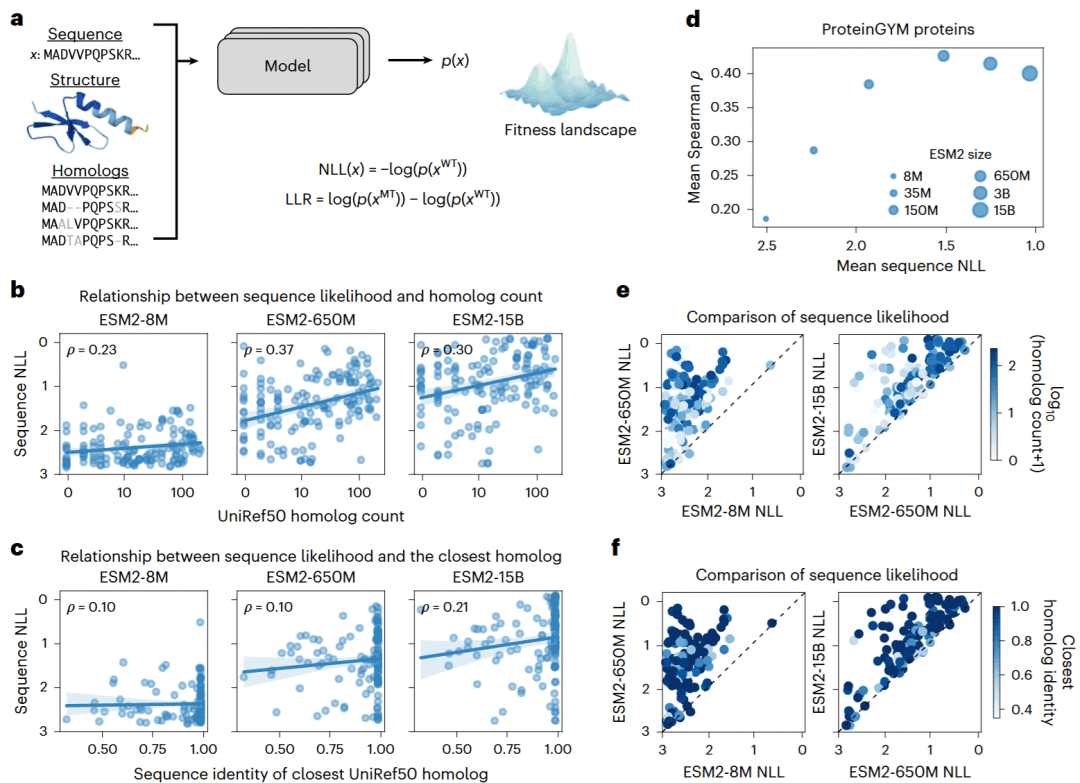
图1: 模型序列可能性的计算方式及其与同源序列数量、模型规模和训练因素的关系。
预测可能性的大小会改变突变分数分布
突变适应度通常通过突变序列与野生型序列之间的对数可能性差计算,因此野生型序列可能性的绝对水平会直接影响所有突变分数。
当模型对野生型残基的置信度很低时,野生型与其他氨基酸的预测概率较为接近,因此所有突变分数都聚集在零附近,模型难以区分中性和有害突变。相反,当模型对野生型残基过度自信时,其他氨基酸的概率接近零,几乎所有突变都会得到强烈负分,同样失去区分能力。
研究人员以抑癌蛋白PTEN为例。小型ESM2模型预测的突变分数主要集中在零附近,而最大模型则把大多数突变预测为强烈有害。中等规模的ESM2-150M更接近实验中观察到的双峰分布,既能识别相对中性的突变,也能识别明显有害的突变,并获得最佳相关性。
在残基层面,ESM2-8M只对极少数残基赋予高置信度,无法完整识别突变敏感位点;ESM2-15B则对几乎所有残基都表现出极高置信度,导致不同位点之间缺乏区分。中等模型预测的残基可能性与实验测得的平均突变效应更一致,也更容易识别PTEN中的癌症热点位点。
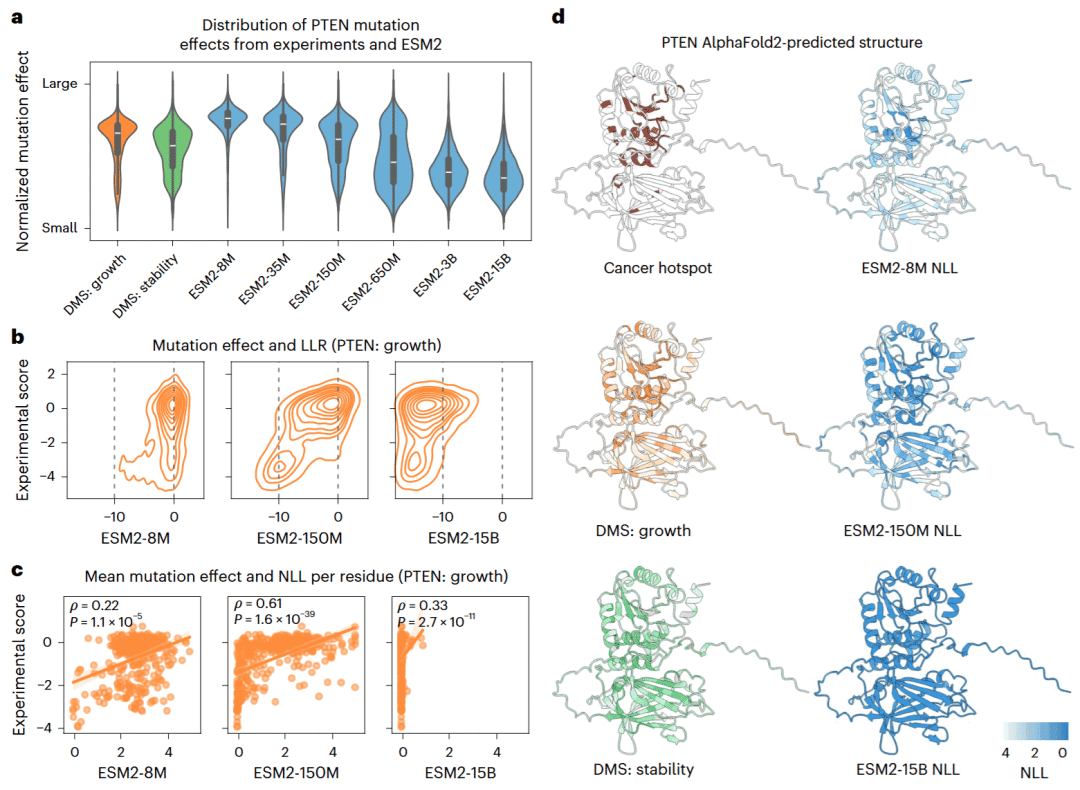
图2: 不同规模ESM2模型对PTEN突变效应分布、残基敏感性和癌症热点的预测结果。
通用模型在中等序列可能性水平表现最佳
研究人员在154项ProteinGYM实验中系统比较野生型序列可能性与适应度预测性能。结果显示,两者呈现明显的钟形关系:当模型预测的序列可能性较低时,性能较差;随着可能性提高,性能逐渐改善;但超过某一水平后,性能再次下降。
对于ESM2系列,平均表现最好的模型是650M参数版本,而不是15B参数版本。最佳性能大致出现在中等野生型序列可能性范围。在这一最佳区间内,不同规模模型的表现相近,说明性能主要由预测可能性所处水平决定,而非参数量本身。
这一钟形趋势不仅存在于ESM2中,也广泛出现在ESM1v、ESMC、CARP、ProGen3和RITA等纯序列模型,以及ESM3、SaProt和ProSST等序列—结构联合模型和ESM-IF1逆折叠模型中。尽管这些模型的架构、训练数据和推断方式不同,它们都在相近的中等序列可能性水平附近取得最佳性能。
对于病毒蛋白和人工设计蛋白,研究人员没有观察到明显的高可能性区性能下降。这是因为这些蛋白质在通用训练集中通常代表不足,模型赋予它们的序列可能性整体偏低,尚未进入过度自信区间。因此,在这类蛋白质上,模型扩展有时仍能持续带来收益。
相比之下,针对单个蛋白质家族训练的位点独立模型和EVE没有表现出明显钟形关系。MSA-Transformer兼具通用与家族信息,其钟形趋势较弱。这提示,非单调规模效应主要是通用模型特有的问题。
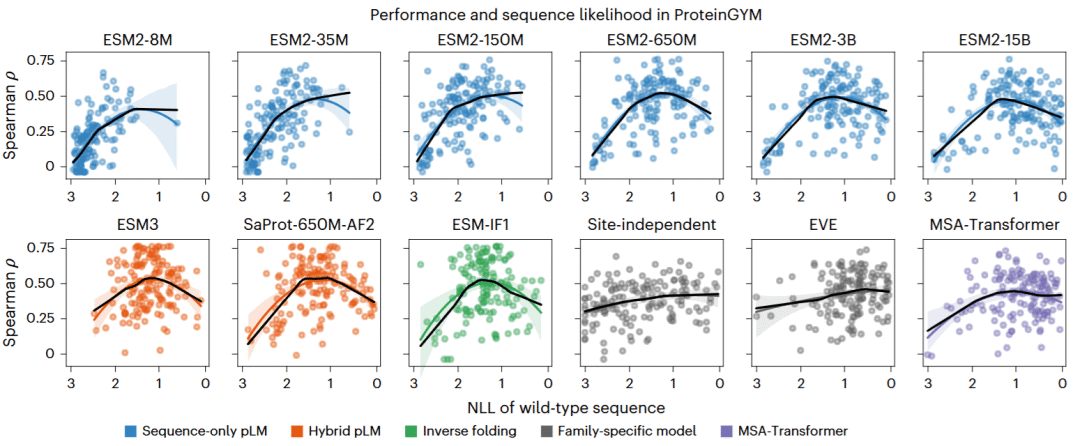
图3 :不同序列、结构和家族特异模型中适应度预测性能与野生型序列可能性的关系。
钟形关系主要源于模型对蛋白质上下文理解的变化
为了进一步解释钟形关系,研究人员将突变预测能力拆分为两个部分。第一部分是上下文理解,即模型能否判断某个残基位置整体上是否容易受到突变影响;第二部分是替换特异性,即在同一个位点上,模型能否区分19种不同氨基酸替换的相对影响。
结果显示,通用模型的上下文理解能力与野生型序列可能性之间呈钟形关系,在中等可能性水平达到最佳。相比之下,替换特异性则随着残基可能性升高而总体单调改善。
这意味着,大模型并非完全失去判断不同氨基酸替换的能力。问题主要在于,模型越大、对野生型序列越自信,就越容易把许多残基都视为高度重要,从而削弱了对不同位点突变敏感性的区分。适应度预测不仅需要知道某一种替换是否合理,也需要正确理解该位点在整个蛋白质结构和功能背景中的重要程度。
家族特异模型在这两个方面都没有表现出明显钟形关系,因为它们直接从同源序列频率和协同变化中学习具体蛋白质家族的约束,较少受到通用模型训练偏差的影响。
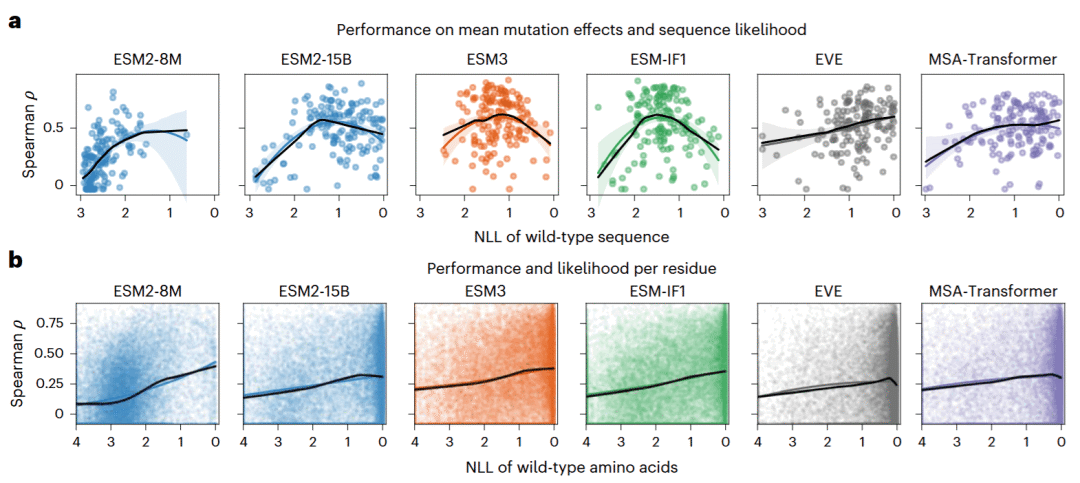
图4: 模型对蛋白质上下文和氨基酸替换特异性的理解与序列可能性之间的关系。
结构环境和突变敏感性揭示不同模型的优势
研究人员利用相对溶剂可及性描述残基的结构环境。通常,埋藏在蛋白质内部的残基更容易因突变而破坏稳定性,而暴露在表面的残基总体更能容忍替换。分析表明,溶剂可及性与实验测得的平均突变效应和有害突变数量之间存在明显关系,尤其在蛋白质稳定性实验中更强。
不同模型预测的高可能性残基通常具有较低溶剂可及性,并包含更多有害突变,说明模型确实能够捕获一部分结构和功能约束。结构信息模型ESM3和ESM-IF1在高置信度区域对结构环境的反映更准确,这与它们直接使用三维结构有关。
然而,在识别真正的突变敏感性方面,家族特异模型表现更好。EVE赋予高序列可能性的蛋白质通常包含更高比例的有害突变,而ESM2-15B的蛋白质序列可能性与有害突变比例之间没有显著关系。这说明通用模型在残基层面可以捕捉一定的保守和结构信息,但把这些信息聚合到整个蛋白质层面时,容易混入较多噪声。
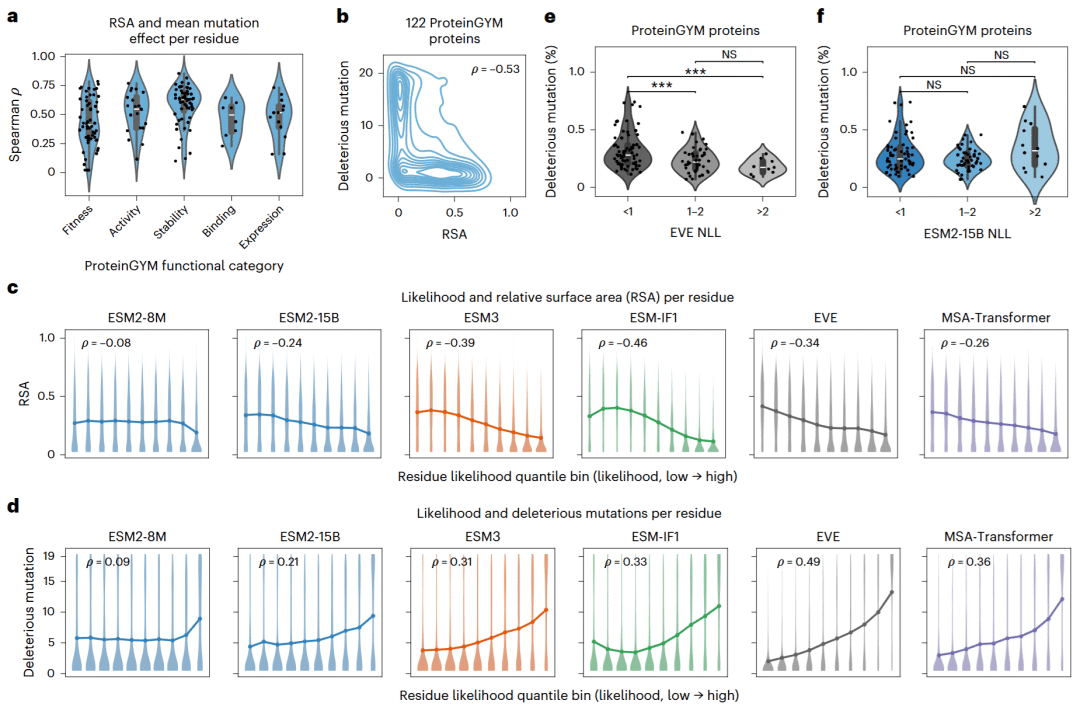
图5: 模型残基可能性与溶剂可及性、有害突变数量和蛋白质突变敏感性的关系。
适应度预测取决于模型是否重现同源序列中的进化模式
家族特异模型直接利用同源序列,因此其残基可能性天然反映进化保守模式。一个残基在同源序列中越保守,模型通常赋予其越高可能性;一个蛋白质中保守位点比例越高,整体序列可能性也越高。
研究人员比较多种模型后发现,不同通用模型之间在残基层面的预测具有一定一致性,家族特异模型之间也高度相关。通用模型与家族特异模型在残基层面存在中等相关,但在蛋白质整体可能性层面相关性很弱,说明逐位聚合会放大噪声。
更重要的是,通用模型的残基可能性与家族特异进化模式越一致,其适应度预测表现越好。模型与位点独立模型、EVE或MSA-Transformer之间的残基可能性相关性,都能预测其在ProteinGYM中的性能。
这种进化一致性本身也与序列可能性呈钟形关系:当模型对野生型序列置信度过低或过高时,与同源序列保守模式的吻合程度都较差;在中等可能性范围内,一致性最高,恰好对应适应度预测性能的峰值。研究人员因此建议,在使用通用模型进行突变效应预测前,先比较其残基可能性与简单同源序列频率模型的一致性。综合覆盖率和性能收益,可将残基级相关系数约0.5作为实用筛选阈值。
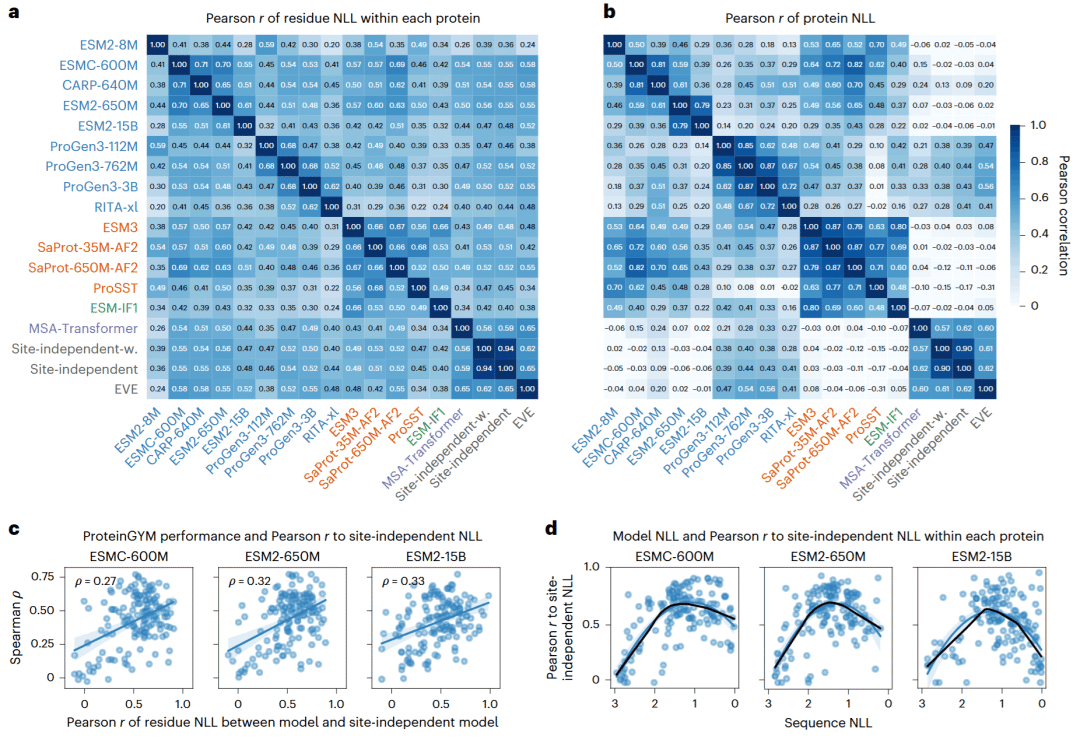
图6: 通用模型与家族特异模型预测的一致性及其与适应度预测性能的关系。
讨论
该研究说明,蛋白质语言模型的规模扩展规律具有明显的任务依赖性。更大的模型通常能够更准确地预测训练分布中的序列,并在结构建模、生成和监督微调任务中表现更好,但零样本适应度预测依赖野生型与突变体之间的相对可能性,因此会受到概率归一化和模型置信度的特殊影响。
当模型尚未充分学习某类蛋白质时,其预测过于平坦,不同突变之间缺乏区分;当模型过度学习或过度自信时,又会把几乎所有偏离野生型的替换都判断为有害。只有在中等置信度下,模型才能较好地重现同源序列中真实的保守与变异模式,从而获得最佳适应度预测性能。
因此,不能简单依据模型参数量、训练损失或序列可能性选择蛋白质适应度预测器。对于零样本预测,应优先考察模型在目标蛋白质上的可能性是否位于合理区间,并评估其残基层面预测是否与同源序列的进化模式一致。对于同源序列丰富的蛋白质,家族特异模型仍然具有重要优势;对于缺乏同源序列、病毒蛋白或人工设计蛋白,通用模型则可能更具价值。
结构信息模型呈现出一定特殊性。它们除了学习进化规律,还直接学习蛋白质的生物物理约束,因此在稳定性相关任务中可能表现更好。未来模型应避免单纯追求更高野生型序列概率,而应分别建模蛋白质上下文、突变敏感性和替换特异性,并通过校准或混合专家策略控制模型置信度。
总体而言,研究人员揭示了蛋白质适应度预测中“大模型不一定更好”的机制:模型规模通过改变序列可能性的绝对水平,间接影响突变分数分布;真正决定性能的是预测是否忠实反映进化和生物物理约束。这一发现不仅适用于蛋白质语言模型,也可能为DNA、RNA语言模型以及基于序列可能性的生物分子设计方法提供参考。
整理 | DrugOne团队
参考资料
Hou, C., Liu, D., Zafar, A. et al. Understanding language model scaling for protein fitness prediction. Nat Comput Sci (2026).
https://doi.org/10.1038/s43588-026-01010-z
内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-13,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号