Nature | 把 RoseTTAFold 变成生成模型:RFdiffusion 的方法细节、实验结果与局限

Nature | 把 RoseTTAFold 变成生成模型:RFdiffusion 的方法细节、实验结果与局限

DrugIntel
发布于 2026-07-13 16:22:21
发布于 2026-07-13 16:22:21

论文题目: De novo design of protein structure and function with RFdiffusion 期刊: Nature 发表时间: 2023 年 核心机构: University of Washington Institute for Protein Design、MIT、Columbia University、Seoul National University 等 DOI: 10.1038/s41586-023-06415-8
导读
RFdiffusion 是 AI 蛋白设计领域非常值得精读的一篇工作。它的核心贡献不是简单把扩散模型搬到蛋白上,而是把 RoseTTAFold 这类结构预测网络改造成蛋白骨架生成器,让模型从随机残基框架出发,逐步去噪生成可设计、可折叠、可实验验证的蛋白结构。更重要的是,它不只生成单体蛋白,还能根据对称性、靶蛋白界面、功能基序和酶活性位点等条件进行定向设计。本文将详细拆解 RFdiffusion 的问题背景、模型表示、扩散过程、条件生成机制、ProteinMPNN 后处理、AlphaFold2 过滤、实验验证结果,并讨论它在真实 AI 制药与蛋白工程中的价值和边界。
为什么这篇论文值得关注?
过去几年,AI 蛋白设计经历了一个很明显的变化:早期重点是预测蛋白会折成什么结构,后来逐渐转向设计一个新序列让它折成目标结构,再进一步走向更难的问题:能否直接设计一个满足功能要求的新蛋白?
这个问题远比生成一个看起来像蛋白的三维结构困难。真实蛋白设计至少要同时满足几类约束:骨架要合理,序列要能编码这个骨架,核心要能稳定折叠,表面要能溶解表达,界面要能形成特异相互作用,如果涉及酶或金属结合,还要把少数关键侧链精准放在空间中的正确位置。
RFdiffusion 值得关注,正是因为它把这些原本分散的问题放进了一个相对统一的生成框架中。作者不是为每一个任务单独写一套规则,而是让一个扩散模型在蛋白残基三维框架上逐步去噪,并通过不同条件信息引导模型走向不同设计目标:无条件生成、指定 fold、指定对称性、指定结合靶点、固定功能基序、构建酶活性中心等。论文摘要中也明确强调,RFdiffusion 的目标是提供一个能够解决多类蛋白设计挑战的通用深度学习框架,而不仅是单一 benchmark 上的模型改进。
研究背景
De novo 蛋白设计的理想目标是从头创造自然界没有的新蛋白,并让它具有预期结构和功能。这里的功能可以是结合一个靶蛋白、稳定呈递一个抗原表位、形成高阶对称纳米结构、固定一个金属离子,或者把几个催化残基摆成酶活性位点。
传统蛋白设计长期依赖物理能量函数、采样搜索和人工经验。Rosetta 体系在规则二级结构、理想化骨架和小型蛋白设计中积累了大量成功案例,但它面临一个根本难题:蛋白构象空间极其巨大,结构稳定性和功能几何约束又高度耦合。如果直接在序列空间或骨架空间里搜索,很容易陷入局部最优,生成速度慢,设计多样性有限。
深度学习带来了新的可能。AlphaFold2、RoseTTAFold 证明了神经网络可以从序列和进化信息中学习非常强的蛋白结构先验。但结构预测模型本质上回答的是给定序列会折成什么,而 de novo 设计需要回答的是怎样生成一个满足目标条件的新结构和新序列。
扩散模型天然适合这个问题。它可以从噪声开始,通过多步去噪逐渐生成复杂样本;每一步还可以加入条件信息,引导生成方向。问题在于,蛋白不是图像。蛋白骨架有严格的几何约束,残基之间存在旋转、平移、局部取向、主链连续性、二级结构和长程相互作用。简单在坐标上加噪声并训练网络去噪,很容易得到不合理骨架或不可设计结构。
RFdiffusion 的关键判断是:与其从零训练一个蛋白扩散模型,不如利用 RoseTTAFold 已经学到的蛋白结构先验,把结构预测网络改造成扩散模型的去噪网络。论文明确指出,RoseTTAFold 适合作为蛋白设计 DDPM 的基础,因为它能高精度生成蛋白结构,采用残基刚体框架表示,具有旋转等变性,并且可以在残基、残基间距离和取向、三维坐标等层面接受条件信息。
这篇论文的核心思想
RFdiffusion 的核心思想可以压缩成一句话:
把 RoseTTAFold 从结构预测网络改造成残基刚体框架上的扩散去噪网络,让模型从随机残基框架逐步生成蛋白骨架,再用 ProteinMPNN 设计序列,并用 AlphaFold2 等工具过滤验证。
这个思想的关键不在扩散模型四个字,而在三层设计:
第一,扩散对象不是氨基酸序列,也不是全原子坐标,而是蛋白主链残基框架。每个残基用 Cα 坐标和 N-Cα-C 定义的刚体取向表示,这样既保留主链三维几何,又避免直接在所有原子自由度上建模带来的复杂性。
第二,去噪网络不是从零训练的小模型,而是预训练 RoseTTAFold。RoseTTAFold 已经学到大量蛋白结构规律,RFdiffusion 通过去噪任务微调它,相当于把结构预测先验转化成生成先验。
第三,生成框架可以被条件化。同一个扩散过程可以输入 fold 信息、固定 motif、靶蛋白表面热点残基、对称性约束等,从而适配不同蛋白设计任务。论文图 1 展示了这一点:RFdiffusion 可以无条件生成,也可以根据对称性、结合靶点、功能基序和对称功能基序生成结构。
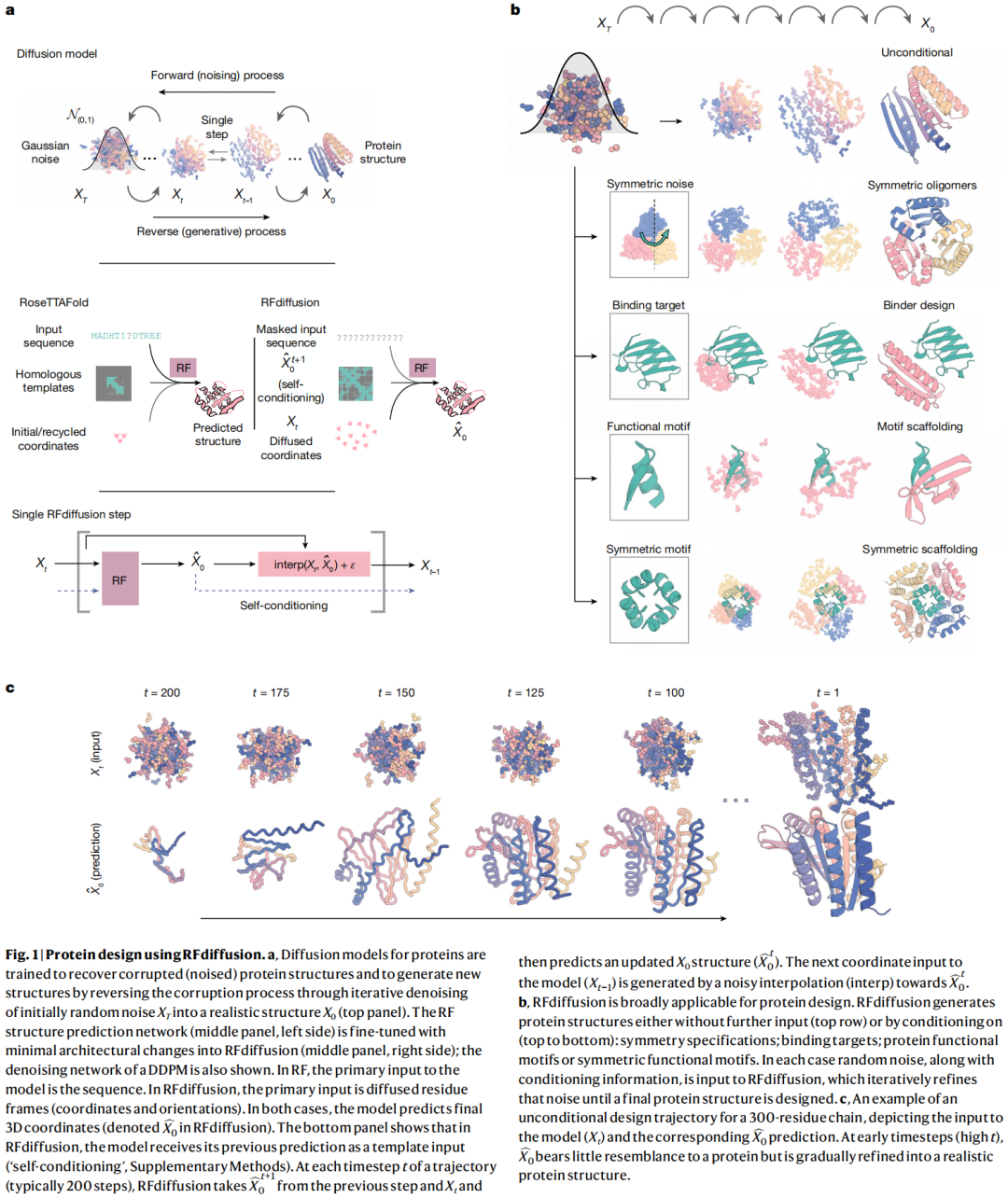
5. 方法细节:RFdiffusion 到底是怎么做的?
5.1 任务定义与输入输出
RFdiffusion 的基本任务是:
给定一个设计条件,或者不给任何条件,从随机噪声出发,生成一个蛋白主链骨架;随后用序列设计模型为这个骨架生成氨基酸序列;再用结构预测和实验手段验证该序列是否能折回设计结构,并实现目标功能。
不同任务的输入略有不同:
- •无条件单体设计: 输入蛋白长度,输出一个新蛋白骨架。
- •fold 条件设计: 输入二级结构和 block adjacency 等 fold 信息,输出满足该拓扑的骨架。
- •对称寡聚体设计: 输入点群对称性和亚基长度,输出满足 C、D、T、O、I 等对称性的组装体。
- •功能基序 scaffold: 输入需要固定的功能残基、主链和侧链几何,输出能够稳定承载该 motif 的新 scaffold。
- •蛋白 binder 设计: 输入靶蛋白结构和界面 hotspot 残基,输出能结合目标位点的新蛋白 binder。
- •酶活性位点 scaffold: 输入少数催化残基的空间构型,输出能固定这些残基的蛋白骨架。
最终输出不是一步到位的可用药物或可用酶,而是一批候选蛋白结构与序列,需要经过 ProteinMPNN 序列设计、AlphaFold2/ESMFold 结构复预测、过滤、表达纯化和功能实验验证。
5.2 数据表示:为什么用残基框架,而不是普通坐标?
RFdiffusion 使用 RoseTTAFold 的 frame representation。每个残基由两部分描述:
- 1.Cα 坐标:表示该残基在三维空间中的位置。
- 2.N-Cα-C 刚体取向:由主链 N、Cα、C 三个原子定义局部坐标系,表示该残基的方向。
这样,一个蛋白骨架就被表示成一串残基刚体框架。这个表示有几个优势:
- • 它比全原子坐标更简洁,降低生成难度。
- • 它保留主链几何的核心信息,适合描述折叠拓扑。
- • 它天然适合旋转等变网络处理,因为整体旋转蛋白时,模型输出也应随之等变。
- • 它可以在残基层面加入条件,如固定某些残基位置、限定 residue-residue 关系、指定界面 hotspot 等。
不足也很清楚:这种表示主要关注主链框架,侧链、质子化状态、金属配位细节、小分子底物和显式溶剂并没有被完整建模。因此,RFdiffusion 生成的是一个强结构先验下的设计起点,而不是对真实分子体系能量面的完整模拟。
5.3 前向扩散:蛋白结构如何被加噪?
训练时,作者从 PDB 中采样真实蛋白结构,并对这些结构进行最多 200 步加噪。对每个残基框架,加噪分为平移和旋转两部分:
- •平移噪声: 对 Cα 坐标加入三维高斯噪声。
- •旋转噪声: 对残基取向在旋转矩阵流形上进行 Brownian motion,也就是在 SO(3) 旋转空间中逐渐扰动方向。
这一步的目标是构造从真实蛋白结构 X0 到高噪声结构 Xt 的前向过程。到高时间步时,残基框架接近随机分布;到低时间步时,结构仍保留较多蛋白形态。
这个设计很重要。蛋白骨架不是普通欧氏空间中的一堆点,残基方向属于旋转空间。如果只对所有坐标简单加噪,模型可能无法正确学习主链局部几何和全局取向之间的关系。RFdiffusion 把平移和旋转分别处理,使扩散过程更符合蛋白骨架的几何结构。论文明确说明,平移部分通过三维高斯噪声扰动 Cα,旋转部分通过旋转矩阵流形上的 Brownian motion 加噪。
5.4 去噪网络:RoseTTAFold 如何变成 RFdiffusion?
RoseTTAFold 原本是结构预测网络。它的典型输入是序列,加上模板、初始坐标等信息,输出蛋白三维结构。
RFdiffusion 对这个逻辑做了反转:
- • 在 RF 中,主要输入是序列,输出是结构。
- • 在 RFdiffusion 中,主要输入是带噪声的残基框架 Xt,输出是预测的干净结构 X0。
- • 对特定任务,还可以输入条件信息,例如固定 motif 坐标、部分序列、fold 信息、靶蛋白结构或热点残基。
也就是说,RFdiffusion 每一步都在回答一个问题:给定当前这个带噪声的蛋白框架,它最可能来自哪个真实、可设计的蛋白骨架?
它并不是预测噪声本身,而是预测最终干净结构 X0。论文图 1 中也强调,每个时间步输入 Xt 和上一轮预测,RFdiffusion 输出更新后的 X0 预测,再通过向该预测插值并加入噪声得到下一步 Xt−1。
5.5 训练目标:为什么 MSE 比 FAPE 更适合扩散?
RoseTTAFold 结构预测常用 FAPE 这类对全局坐标不敏感的损失,因为结构预测关心的是相对几何是否正确,而不关心蛋白整体放在哪个坐标系里。
但扩散生成不同。反向去噪需要在连续时间步之间保持全局坐标轨迹的一致性。如果损失函数完全不关心全局参考系,模型每一步预测的结构可能在坐标系中跳动,不利于稳定地从 Xt 走到 Xt−1。
因此,RFdiffusion 使用的是预测残基框架与真实蛋白结构之间的均方误差损失,并且不做对齐。这个 MSE 在所有残基上平均。作者明确指出,与 FAPE 不同,MSE 不具有全局参考系不变性,因此能促进时间步之间全局坐标框架的连续性。消融实验也显示,MSE 对无条件生成非常关键。
这说明 RFdiffusion 的训练目标不是随便套用扩散模型标准做法,而是围绕蛋白结构去噪的几何连续性做了调整。
5.6 Self-conditioning:为什么上一轮预测也要喂给模型?
RFdiffusion 还使用了 self-conditioning。简单说,模型在当前时间步不仅看到带噪声结构 Xt,也会看到上一时间步对干净结构的预测。这个思想类似 AlphaFold2 和 RoseTTAFold 中的 recycling:模型反复使用之前的结构估计来修正当前判断。
在扩散生成中,这样做可以让去噪轨迹更连贯。每一步预测不再是孤立估计,而是在前一次结构假设基础上继续更新。论文消融显示,self-conditioning 显著提升无条件和条件蛋白设计任务的 in silico 表现;去掉 self-conditioning 后,模型虽然能生成一些二级结构,但核心 packing 和整体理想性明显变差。
这个细节非常重要。很多人理解扩散模型时只关注从噪声到样本,但在蛋白这种几何约束极强的对象上,轨迹稳定性本身就是模型能力的一部分。
5.7 反向生成:从随机残基框架到蛋白骨架
推理时,RFdiffusion 先初始化一组随机残基框架 XT。然后重复以下过程:
- 1. 把当前带噪声残基框架 Xt 输入 RFdiffusion。
- 2. 模型预测一个干净蛋白结构 X0。
- 3. 根据扩散反向过程,将 Xt 向预测的 X0 方向移动一步。
- 4. 加入适量噪声,得到 Xt−1。
- 5. 重复去噪,直到得到最终骨架。
早期时间步中,模型预测的 X0 还不像真实蛋白,因为随机残基框架对应的可能蛋白结构范围太宽。随着去噪推进,可能结构范围逐渐收窄,预测开始出现二级结构、核心 packing 和清晰拓扑。论文图 1c 就展示了一个 300 残基蛋白从 t=200 到 t=1 的生成轨迹:早期杂乱,后期逐渐形成清晰蛋白折叠。
值得注意的是,RFdiffusion 在每一步都预测最终结构,因此推理时可以采用较大步长或提前截断轨迹来提高效率。论文报告 100 残基蛋白最快可约 11 秒生成,这对后续批量设计很重要。
5.8 条件生成:同一个去噪框架如何适配不同任务?
RFdiffusion 的强大之处在于条件接口非常灵活。
指定 fold
对于 TIM barrel、NTF2 等拓扑,模型可以接收二级结构和 block adjacency 信息。二级结构告诉模型哪些区域应形成 helix 或 strand;block adjacency 告诉模型这些二级结构片段在空间上如何相邻。这样,模型不是完全自由生成,而是在给定拓扑框架内采样多样骨架。
指定对称性
对于对称寡聚体,输入包括点群对称性和单体长度。模型先为一个亚基生成随机残基框架,再复制出 n−1 个亚基并按指定点群排列。由于 RFdiffusion 继承了 RF 的旋转和链重标号等变性,对称结构在去噪过程中大体可以保持;作者还在每一步显式重新对称化。对于八面体和二十面体等大组装体,只显式建模生成完整组装体所需的最小亚基集合,以降低计算和显存成本。
固定功能基序
对于 motif scaffolding,输入是必须保留的功能片段,包括主链坐标、侧链和序列。模型在去噪过程中生成新的 scaffold,同时让这些关键残基保持在正确空间位置。这对于酶活性位点、蛋白相互作用片段、金属配位残基和表位呈递非常关键。
设计 protein binder
对于 binder 设计,输入是靶蛋白结构和目标界面 hotspot 残基。Hotspot 的作用是告诉模型应该结合靶蛋白的哪个区域,而不是让 binder 随机贴到表面任意位置。Extended Data Fig. 8 说明,指定 hotspot 可以显著改变 binder 的目标位点,并能把设计从一个优先结合位点重定向到另一个位点。
外部势能引导
在某些任务中,作者还加入外部 potential 进行引导。例如对称寡聚体设计中使用链内和链间 contact potential,使亚基更紧凑并推动链间接触,从而提高 in silico 成功率。Extended Data Fig. 5 显示,这类外部势能可以显著改善对称设计结果。
5.9 序列设计与过滤:RFdiffusion 不是单独完成全部设计
RFdiffusion 主要生成蛋白骨架。生成骨架后,作者使用 ProteinMPNN 为骨架设计序列,通常每个设计采样 8 条序列。作者也考虑过在 RFdiffusion 内同时设计结构和序列,但由于骨架生成加 ProteinMPNN 的组合效果已经很好,没有深入探索联合生成。
随后,作者用 AlphaFold2 或 ESMFold 进行结构复预测。核心逻辑是:如果 ProteinMPNN 设计出的序列真的能编码 RFdiffusion 生成的骨架,那么 AF2 从单序列预测出的结构应该与设计模型高度一致。
论文中的通用 in silico 成功标准相当严格:
- • AF2 预测高置信度,mean pAE < 5;
- • AF2 预测结构与设计结构的全局 backbone RMSD < 2 Å;
- • 如果有 scaffolded functional site,该功能位点 backbone RMSD < 1 Å。
作者认为这个标准比一些基于 TM-score 的指标更严格,并且与已有实验成功结果具有相关性。
对于 binder 设计,过滤还会关注 AF2 monomer pLDDT、interaction pAE、binder 单体与设计结构的 RMSD 等指标。Extended Data Fig. 8 中,binder 的 in silico success 定义为 AF2 monomer pLDDT > 80、AF2 interaction pAE < 10、AF2 monomer versus design RMSD < 1 Å。
5.10 方法流程小结
RFdiffusion 的完整工作流可以概括为:
- 1.准备设计条件:蛋白长度、目标 fold、对称性、功能 motif、靶蛋白结构、hotspot 残基等。
- 2.初始化随机残基框架:每个残基由 Cα 坐标和 N-Cα-C 局部取向表示。
- 3.RFdiffusion 多步去噪生成骨架:RoseTTAFold 微调后的去噪网络不断预测干净结构,并更新残基框架。
- 4.施加条件和引导:固定 motif、保持对称性、指定靶点界面、必要时加入 contact potential。
- 5.ProteinMPNN 设计序列:为生成骨架采样能够编码该结构的氨基酸序列。
- 6.AF2/ESMFold 复预测过滤:检查序列是否能折回设计骨架,binder 是否可能形成目标界面。
- 7.实验验证:表达纯化、CD、SEC、BLI、YSD、nsEM、cryo-EM 等验证结构和功能。
6. 实验设计与关键结果
6.1 无条件单体生成:从噪声生成可折叠蛋白
RFdiffusion 可以在没有任何条件输入的情况下生成多样蛋白单体,覆盖 alpha、beta 和 alpha/beta 混合拓扑。AF2 和 ESMFold 对许多设计的复预测结构与 RFdiffusion 设计模型非常接近,论文中展示了 300 和 600 残基设计的例子。作者还发现,随着蛋白长度增加,生成结构与 PDB 中已知结构的最高 TM-align 相似性下降,说明模型不是简单记忆训练集结构。
实验上,作者表征了 6 个 300 aa 设计和 3 个 200 aa 设计,圆二色谱显示其二级结构与设计模型一致,并且具有很高热稳定性。图 2 还显示,去掉预训练、去掉 RFdiffusion fine-tuning、去掉 MSE loss 或去掉 self-conditioning 都会明显降低性能,说明 RFdiffusion 的成功不是单一模块带来的,而是结构预测先验、扩散去噪目标、全局坐标连续性和自条件共同作用的结果。
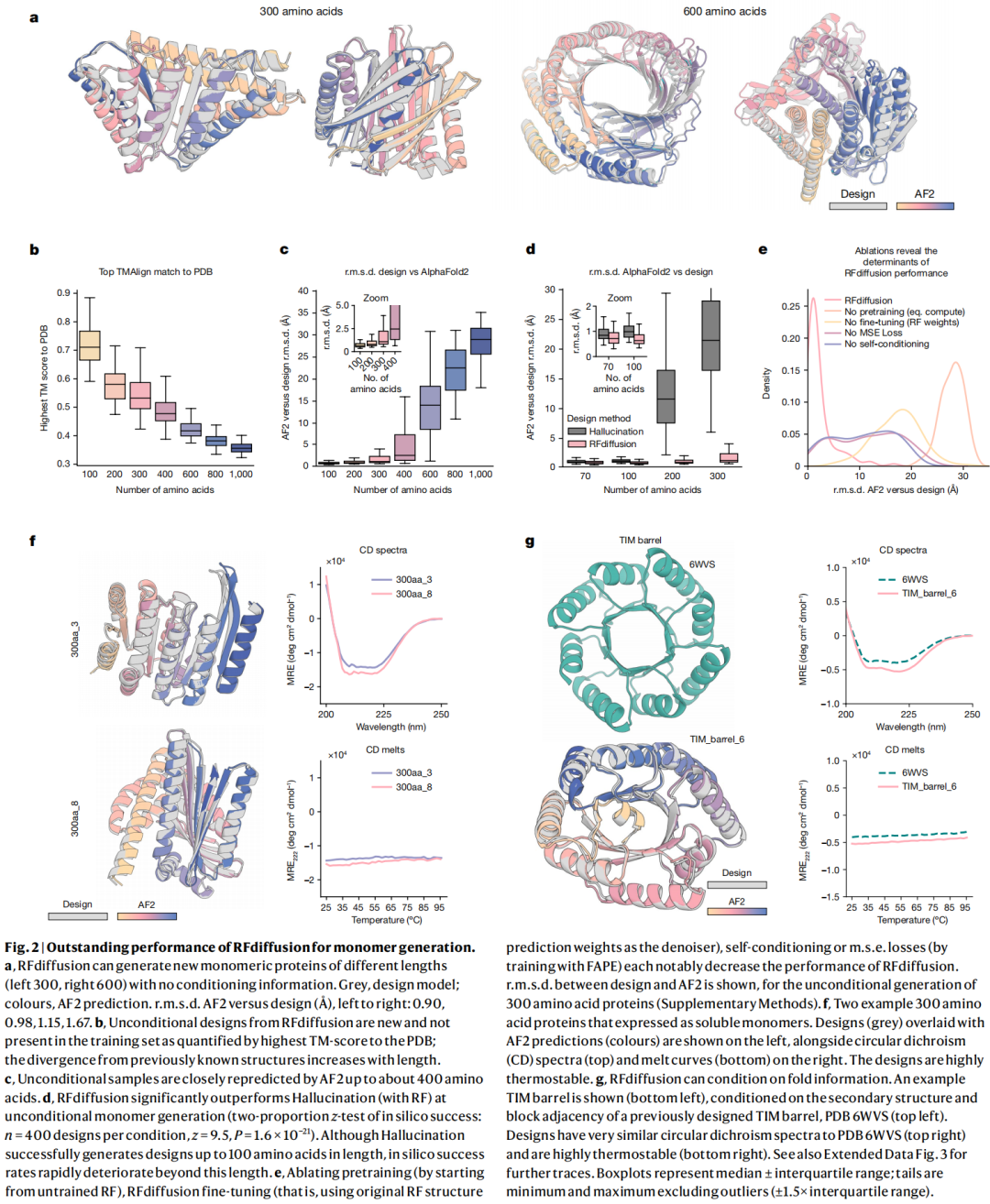
6.2 Fold 条件设计:让模型按指定拓扑生成
作者进一步微调模型,使其可以根据二级结构和 fold 信息生成指定拓扑。TIM barrel 和 NTF2 是两个典型案例,分别与酶设计、小分子结合蛋白设计有关。论文报告 TIM barrel 和 NTF2 fold 的 in silico 成功率分别为 42.5% 和 54.1%;对 11 个 TIM barrel 设计进行实验表征,至少 8 个可溶、热稳定,并且 CD 谱与设计模型一致。
这个结果说明,RFdiffusion 不只是能生成蛋白样结构,还能在用户指定的拓扑空间中进行采样。这一点对功能设计很关键,因为很多功能并不是任意骨架都能承载,而是需要特定几何环境。
6.3 对称寡聚体设计:从单体生成到高阶组装
对称蛋白组装体在疫苗展示、递送载体、纳米材料和催化中都有价值。RFdiffusion 可以生成多种点群对称结构,包括 cyclic、dihedral、tetrahedral、octahedral 和 icosahedral 等。作者对 608 个设计进行实验表征,SEC 显示至少 87 个设计的寡聚状态与模型接近,若按 99% 置信区间则为 126 个。多个设计进一步通过 nsEM 或 cryo-EM 观察到与设计模型一致的二维分类和三维重构。

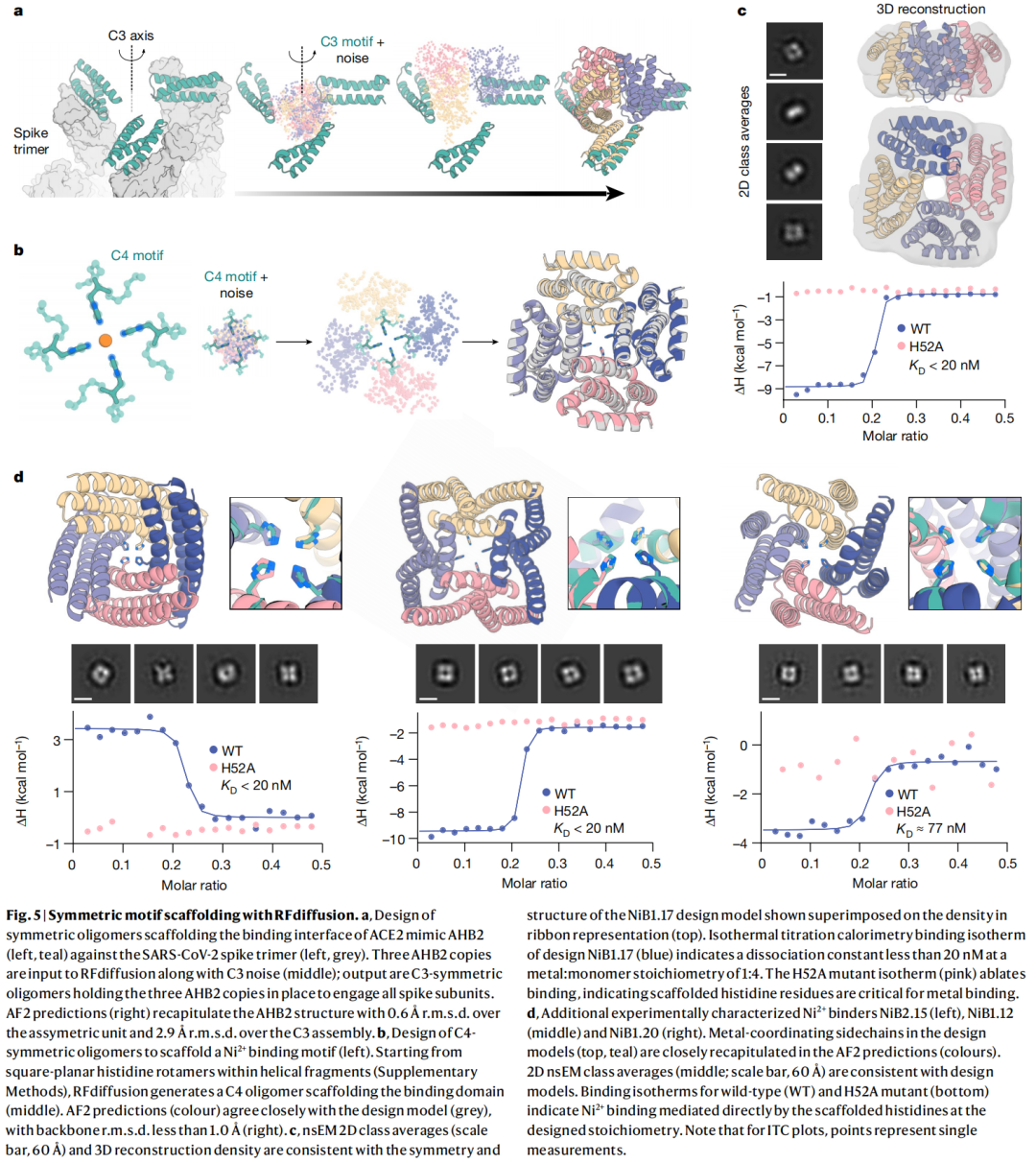
这一部分的意义不只在成功率。更重要的是,RFdiffusion 可以生成自然界少见甚至 PDB 中未见的全局组装拓扑,例如扩展 TIM barrel 样的 C6、C8 alpha/beta barrel。这说明深度生成模型在蛋白组装设计中可能探索到自然演化没有充分采样的结构区域。
6.4 功能基序 scaffold:把关键残基固定到正确位置
功能基序 scaffold 是蛋白设计中非常难的一类任务。它要求模型不是随便生成稳定蛋白,而是围绕少数关键残基构建一个能支撑其三维构象的稳定骨架。
作者构建了包含 25 个 motif-scaffolding 问题的 in silico benchmark,来源于 6 篇近期工作,覆盖病毒表位、受体 trap、小分子结合位点、蛋白界面和酶活性位点等。RFdiffusion 解决了其中 23 个问题,而 Hallucination 解决 15 个,RFjoint Inpainting 解决 19 个;在 23 个已解决问题中,有 19 个 RFdiffusion 的成功设计比例高于另外两类方法。
一个代表性实验是 p53 helix-MDM2 体系。作者使用针对蛋白复合物微调的 RFdiffusion,在 MDM2 存在下 scaffold p53 helix,使设计 scaffold 不仅固定 p53 helix,还能与 MDM2 形成额外接触。最终 96 个设计中有 55 个在 10 μM 下显示可检测结合,其中两个 binder 的亲和力达到 0.5 nM 和 0.7 nM,而 p53 peptide 本身报道亲和力约为 600 nM。
这个结果很有说服力,因为它不是只证明结构能复现 motif,而是证明 scaffold 后的功能可以明显增强。
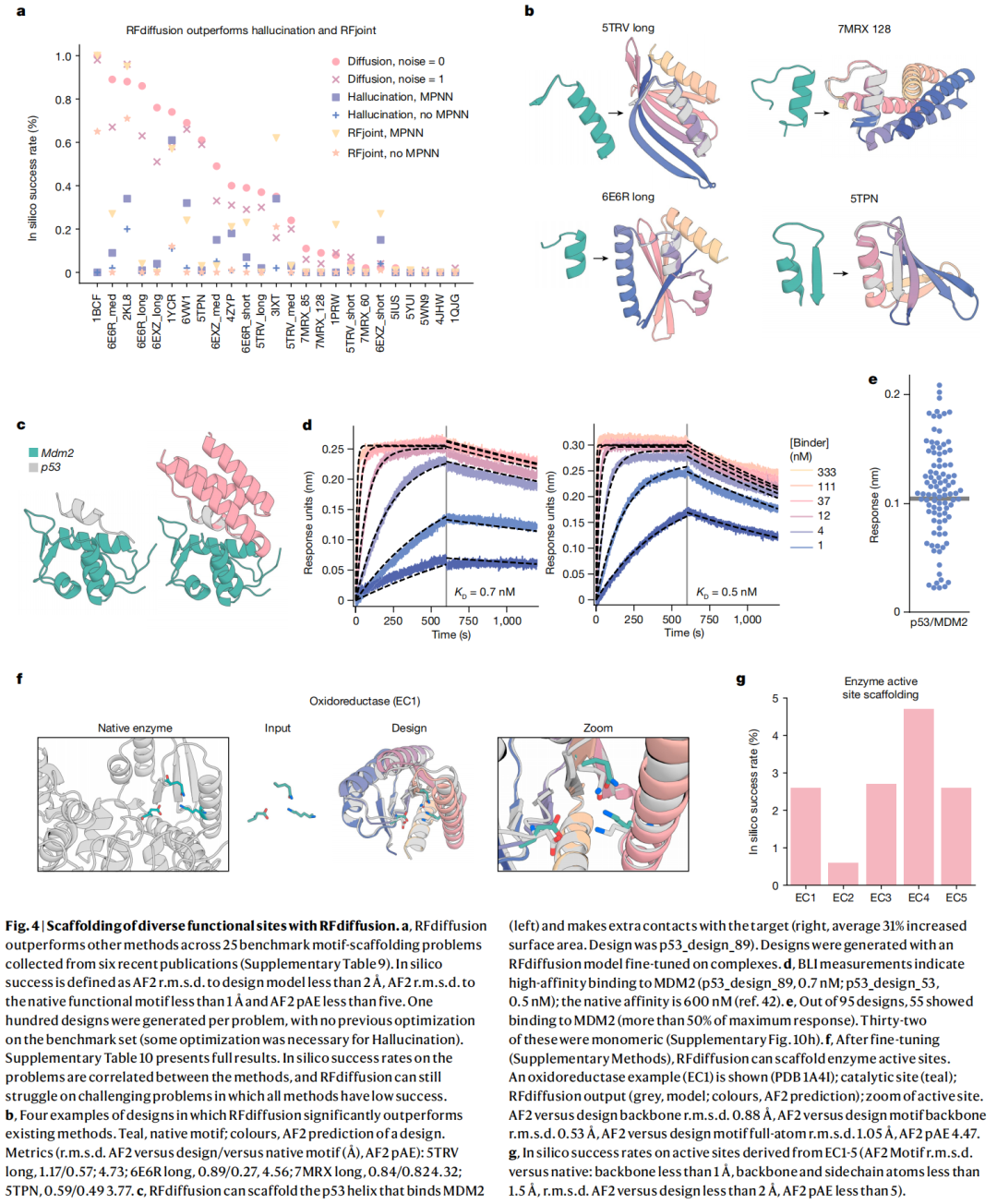
6.5 酶活性位点 scaffold:仍是难题,但方向清晰
酶设计要求更高,因为催化功能往往依赖几个残基、底物、过渡态、局部电场和动态构象的精确配合。RFdiffusion 在微调后可以 scaffold 多类酶活性位点,覆盖 EC1-EC5 的不同酶类别。论文展示了氧化还原酶活性位点 scaffold 示例,AF2 复预测与设计模型和 motif 都高度接近。
但这里也暴露了限制:RFdiffusion 当前不能显式建模结合的小分子底物。作者用外部 potential 隐式引导模型在活性位点周围生成 pocket,并以 retroaldolase 活性位点为示例。这说明 RFdiffusion 可以帮助搭建几何 scaffold,但真正的酶催化设计仍需要更强的化学、反应坐标和底物显式建模。
6.6 De novo binder 设计:从结构指定到实际结合
RFdiffusion 最接近 AI 制药应用的部分,是 de novo protein binder 设计。作者对 5 个靶标设计 binder:Influenza A H1 HA、IL-7Rα、InsR、PD-L1 和 TrkA。所有靶标都找到了实验结合命中。对于 IL-7Rα、InsR、PD-L1 和 TrkA,RFdiffusion 加 AF2 过滤的实验成功率比原始设计流程高约两个数量级;作者估计其中一个数量级来自 RFdiffusion 本身,另一个来自 AF2 过滤。
最有代表性的案例是 influenza HA binder HA_20。HA_20 的 BLI 测得 KD 为 28 nM。更关键的是,作者解析了 HA/HA_20 复合物 2.9 Å cryo-EM 结构,实验结构与计算设计模型高度一致,复合物 RMSD 为 0.63 Å,binder 单独结构相对设计模型偏差约 0.6 Å。
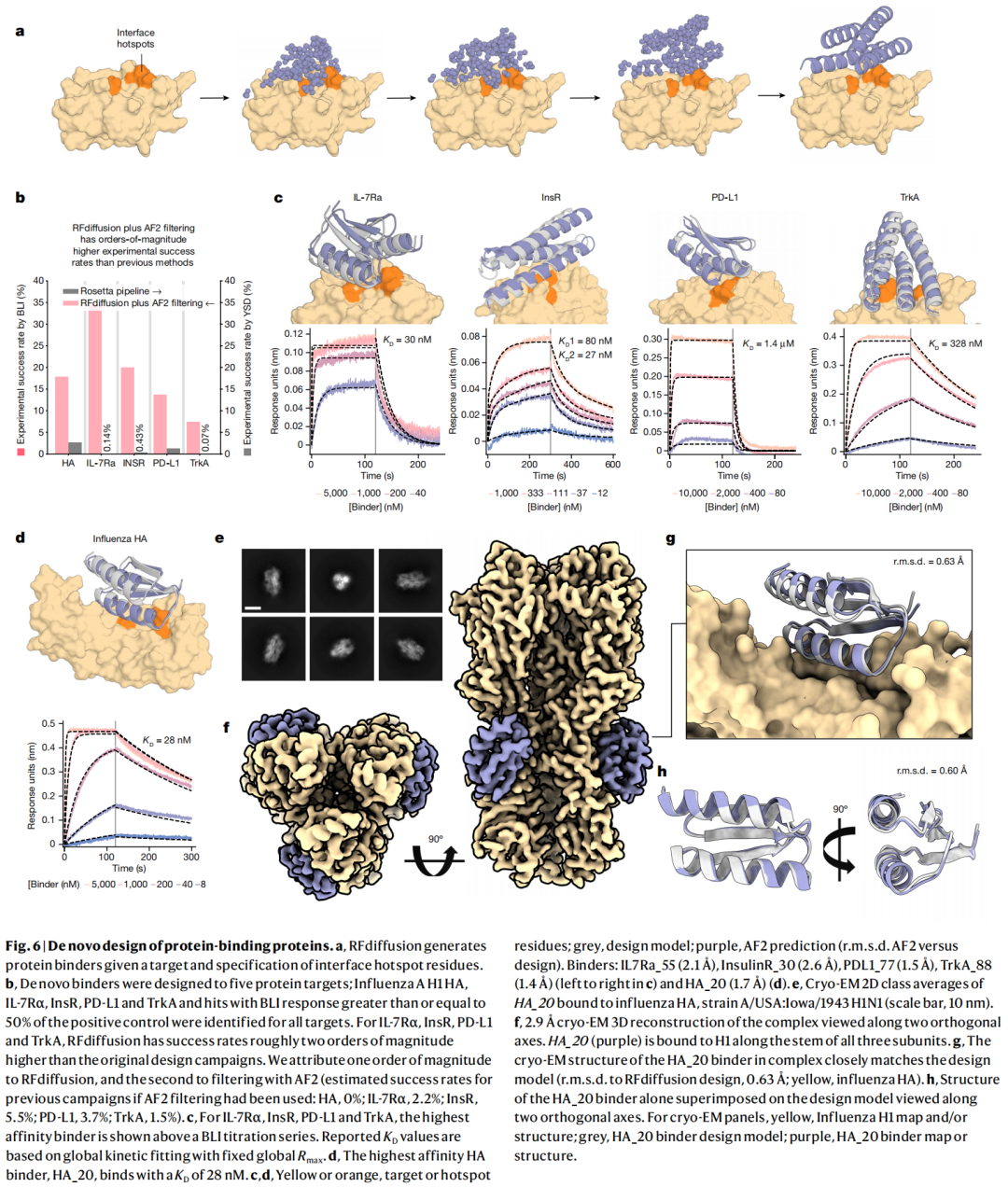
这类结果对领域影响很大。它表明 RFdiffusion 不只是生成可折叠蛋白,还能在重要治疗靶点上设计具有原子级结构准确性的结合蛋白。
启发
启发一:结构预测网络可以成为生成模型的先验
RFdiffusion 最值得学习的一点,是没有把生成模型和结构预测模型割裂开来。RoseTTAFold 已经学到了蛋白结构规律,RFdiffusion 把这种结构知识转化为扩散去噪能力。这比从零训练一个蛋白生成模型更高效,也更符合蛋白设计数据有限、几何复杂的现实。
启发二:蛋白设计的关键不只是生成,而是条件控制
无条件生成蛋白很重要,但真正有应用价值的是围绕目标功能进行设计。RFdiffusion 的条件接口让模型可以根据 symmetry、fold、motif、hotspot 和 target structure 进行生成,这使其从结构生成工具变成更接近功能设计平台的工具。
启发三:AI 蛋白设计从搜索问题转向采样问题
传统设计更像在巨大空间里搜索一个满足约束的点。RFdiffusion 则更像学习一个可设计蛋白结构分布,并从条件分布中采样。这种变化会影响整个工作流:未来设计者可能更多是在定义分子规格、设置约束、采样候选、过滤验证,而不是手工调整每个结构片段。
启发四:实验验证仍然是判断生成模型价值的核心
这篇文章真正有分量,不只是因为模型表现好,而是因为作者做了大量实验验证,包括 CD、SEC、BLI、YSD、nsEM 和 cryo-EM。尤其 HA_20 的 2.9 Å 复合物结构与设计模型高度一致,为 RFdiffusion 的原子级设计能力提供了强证据。
局限性
1. RFdiffusion 主要设计骨架,序列和功能仍依赖后续模型与实验
RFdiffusion 生成的是蛋白骨架,序列设计主要交给 ProteinMPNN,结构复预测依赖 AF2/ESMFold。这种模块化流程非常实用,但也意味着模型没有真正端到端学习结构、序列、侧链相互作用和功能之间的完整耦合。对于普通稳定蛋白,这可能已经足够;但对于酶催化、变构调控、低亲和弱相互作用和高度极性界面,骨架先行可能并不足以保证功能。
2. 对小分子、底物和化学反应的显式建模不足
论文也承认,RFdiffusion 当前不能显式建模结合的小分子底物,只能通过外部 potential 隐式诱导活性位点周围的 pocket。 这对酶设计尤其关键。真正的催化设计需要考虑底物结合姿态、过渡态稳定、质子转移、局部介电环境、金属配位和动态重排。RFdiffusion 能帮助搭 scaffold,但还不是完整的酶设计解决方案。
3. AF2 过滤强大,但也可能带来评价闭环偏差
RFdiffusion 使用 AF2/ESMFold 作为设计验证工具,这是当前非常有效的实践。但需要注意,RFdiffusion 本身源自结构预测网络,ProteinMPNN、AF2 和 RFdiffusion 都在某种程度上学习 PDB 结构分布。若评价主要依赖 AF2 复预测,可能存在模型体系内部相互认可的问题。最终仍需要实验结构和功能数据来确认设计是否真实有效。
4. 实验成功率已经很高,但不是所有任务都同等成熟
RFdiffusion 在非极性目标位点上的 binder 设计表现非常强,但论文讨论中也指出,还需要进一步研究其在更极性靶点、没有天然结合伙伴的位点上的成功率。 对真实药物发现而言,很多靶点表面平坦、柔性强、极性大、构象多态明显,这些场景可能比论文中的部分 benchmark 更难。
5. 蛋白柔性和细胞环境仍然简化
RFdiffusion 主要基于静态结构设计。真实蛋白在溶液中存在构象集合,靶点也可能发生诱导契合、局部重排、糖基化、膜环境限制、竞争性结合和细胞内降解。一个静态复合物模型与实验结构接近,并不一定意味着它在体内有理想药效、稳定性、免疫原性和药代性质。
6. 降低采样噪声提高成功率,但可能牺牲多样性
Extended Data Fig. 1 显示,降低反向过程中的噪声 scale 可以提高 in silico 成功率,但会降低设计多样性。 这反映了生成模型常见的质量-多样性权衡。真实项目中,如果过度追求 AF2 通过率,可能得到一批相似候选;如果追求多样性,又会增加实验筛选成本。
对 AI 制药未来发展的意义
1. 蛋白药物设计会更接近结构驱动的生成流程
RFdiffusion 让 de novo binder 设计从大规模筛选逐渐转向结构条件生成。过去设计一个 binder 可能需要测试成千上万甚至更多序列;论文认为 RFdiffusion 结合改进过滤,可以把高亲和 binder 的发现压缩到几十个设计量级。 这对抗体替代 scaffold、细胞因子调节、受体阻断、病毒中和蛋白等方向都有启发。
2. AI 制药中的蛋白设计比小分子生成更快走向实验闭环
小分子设计要同时处理合成可及性、ADMET、构象、靶点柔性、打分函数偏差和药化优化。蛋白设计也很难,但蛋白结构预测、序列设计和表达验证已经形成较强工具链。RFdiffusion 的成功说明,在结构生物学数据和强预测模型支持下,蛋白设计更容易建立生成-过滤-实验验证的闭环。
3. 生成式模型的价值在于降低提出可测假设的成本
RFdiffusion 并不消除实验,也不替代生物学判断。它真正改变的是候选产生方式:从人工构思少量 scaffold,变为根据明确分子规格批量生成可测试结构。对药物发现而言,这相当于降低了提出高质量蛋白干预假设的成本。
4. 下一代模型需要走向显式多组分建模
未来 RFdiffusion 类模型很可能继续扩展到蛋白-小分子、蛋白-核酸、蛋白-糖链、蛋白-膜环境和多状态构象设计。论文讨论也提到,RFdiffusion 的范围可继续扩展,例如结合其他分子类型、进行 partial noising/denoising 的结构 refinement 等。 对 AI 制药而言,真正理想的模型不是只生成稳定蛋白,而是能在多组分分子系统中同时处理结构、亲和力、特异性、动力学和可开发性。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-13,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号