生成化学:深度学习生成模型驱动的药物发现

生成化学:深度学习生成模型驱动的药物发现

DrugIntel
发布于 2026-06-24 14:06:19
发布于 2026-06-24 14:06:19

文献来源:Bian, Y. & Xie, X.-Q. (2021). Generative chemistry: drug discovery with deep learning generative models. Journal of Molecular Modeling, 27, 71. DOI:https://doi.org/10.1007/s00894-021-04674-8 作者单位:匹兹堡大学药学院药学系 & 计算化学基因组学筛选中心
摘要速览
传统药物研发面临成本高企(平均 28 亿美元)、周期漫长(逾 12 年)的结构性困境,且这些数字仍在持续攀升。本综述系统梳理了以深度学习生成模型为核心的"生成化学"范式,覆盖从化学信息学基础设施到四类前沿生成架构(RNN、VAE、AAE、GAN)的完整技术图谱,并展望了该领域的挑战与未来方向。
一、背景:从传统计算化学到生成化学的范式跃迁
1.1 传统虚拟筛选方法的局限
药物发现中的先导化合物识别,长期依赖两大路线:
- • 基于结构的方法(Structure-Based):以靶蛋白三维结构为基础,涵盖分子对接(Molecular Docking)、分子动力学模拟(MD Simulation)及片段生长(Fragment-Based)策略。
- • 基于配体的方法(Ligand-Based):以已知活性分子为出发点,包括药效团建模(Pharmacophore Modeling)、骨架跃迁(Scaffold Hopping)和分子指纹相似性搜索。
机器学习的引入进一步丰富了虚拟筛选(VS)管线,使得对海量化合物库的高通量评估成为可能。然而,传统方法本质上是在已知化学空间中检索,而非主动创造新结构——这正是生成化学的核心突破所在。
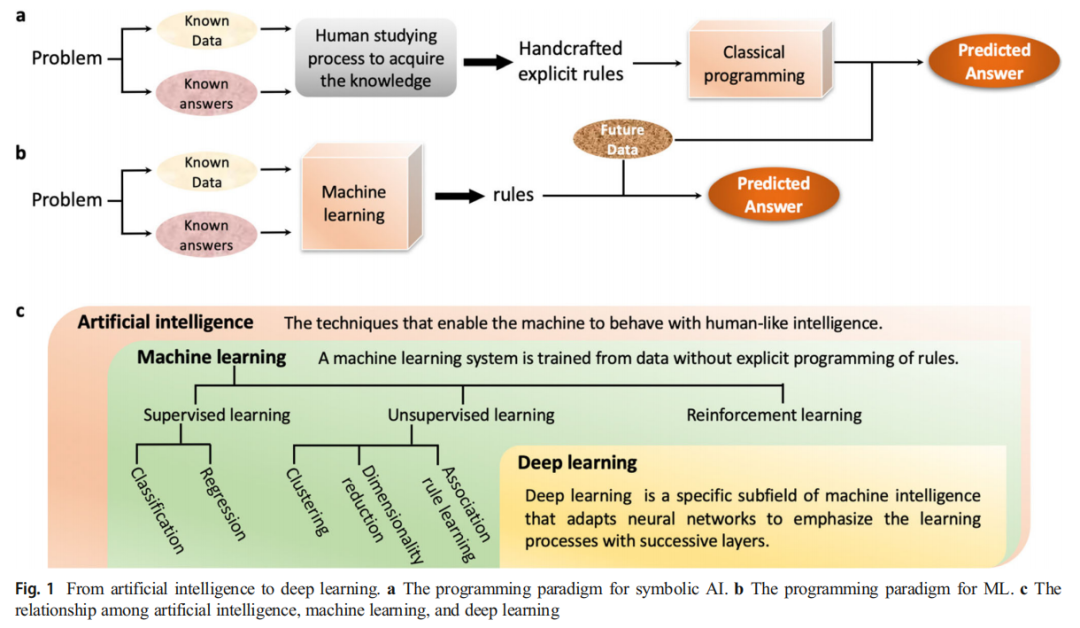
1.2 人工智能在药物发现中的演进
时代 | 代表方法 | 核心范式 |
|---|---|---|
符号 AI 时代 | 专家规则系统(如 Lipinski 五规则) | 人工编码规则 → 预测答案 |
机器学习时代 | SVM、随机森林、朴素贝叶斯 | 数据 + 标签 → 自动归纳规则 |
深度学习时代 | CNN、RNN、GNN | 端到端表示学习,多层特征抽象 |
生成 AI 时代 | VAE、GAN、AAE | 从分布中采样,从零创造新结构 |
深度学习(DL)作为机器学习的特殊子领域,其核心优势在于表示学习——通过多层神经网络将原始输入(如 SMILES 字符串、分子图)逐层转化为越来越抽象的隐表示,从而支持复杂的分类、回归乃至生成任务。
二、基础设施:化学信息学数据库与分子表示
2.1 核心化学数据库
生成化学模型的训练质量高度依赖数据来源,以下为本文梳理的主要数据库:
数据库 | 规模 | 核心用途 |
|---|---|---|
PubChem | ~1.03 亿化合物 | 综合化学信息,NMR谱、理化性质、生物互作 |
ChEMBL | ~200 万类药分子,13,000 个靶点 | 生物活性数据,配体导向药物发现 |
DrugBank | ~14,000 个药物 | 已批准药物及靶点信息,药物再定位研究 |
ZINC | ~2.3 亿可购买化合物 | 虚拟筛选,提供 ready-to-dock 格式 |
RCSB PDB | 大分子三维结构 | 分子对接、热点识别、MD 模拟 |
ASD | 变构调节剂 | 变构药物研究 |
GDB | 组合化学生成库 | 拓展化学空间覆盖 |
趋势:靶标特异性化学基因组学数据库(如 Pain-CKB)正在兴起,将化学、基因与蛋白信息整合,支持系统药理学研究。
2.2 分子表示方式
分子表示(Molecular Representation)是连接化学结构与机器学习算法的关键桥梁:

字符串类表示
- • SMILES:基于 ASCII 字符的线性表示,人类可读,天然适配序列生成模型(如 RNN)。同一分子可生成多种 SMILES,需规范化(Canonicalization)处理。
- • InChI / InChI Key:提供互变异构体不变的标准化表示,常用于去重。
指纹类表示
- • ECFP(扩展连通性指纹):以每个重原子为中心,半径递增地枚举子结构环境,生成定长位向量,适配 ML 分类器(SVM、RF 等)。
- • MACCS Keys:166 位结构键,每位对应特定子结构 SMARTS 模式。
- • Path / Tree Fingerprints:基于线性或树状片段枚举。
⚠️ 指纹的关键局限:不可逆——无法从指纹直接重建分子结构,限制了其在端到端生成模型中的应用。
图类表示
- • 分子图(Molecular Graph):节点对应原子(含原子类型的 One-Hot 编码),边对应化学键(含键类型权重)。可直接供图神经网络(GNN)处理,保留完整拓扑信息。
张量类表示
- • 将原子类型、键类型与连接信息编码为多维张量,适配 CNN 架构。
2.3 工具链
类别 | 代表工具 | 主要功能 |
|---|---|---|
化学信息学 | RDKit、Open Babel、CDK | 格式转换、子结构搜索、指纹计算、描述符生成 |
工作流 | KNIME | 可视化流程编排,批量化合物处理 |
深度学习框架 | TensorFlow、PyTorch、Keras | 神经网络构建与训练 |
经典 ML | Scikit-Learn | SVM、随机森林、逻辑回归等 |

三、四大生成架构详解
3.1 循环神经网络(RNN / LSTM)
原理
RNN 具有内部循环状态 h,能够处理序列数据并在序列元素间传递信息,输出计算公式为:
y = activation(Wo·x + Uo·h + bo)标准 RNN 受梯度消失问题困扰,难以捕获长程依赖。LSTM(长短时记忆网络) 通过引入携带轨道(Carry Track)c 解决了这一问题:
y = activation(Wo·x + Uo·h + Vo·c + bo)LSTM 的遗忘门、输入门与输出门协同控制信息的保留、更新与输出,使模型能够在长序列中维持有效的梯度流。
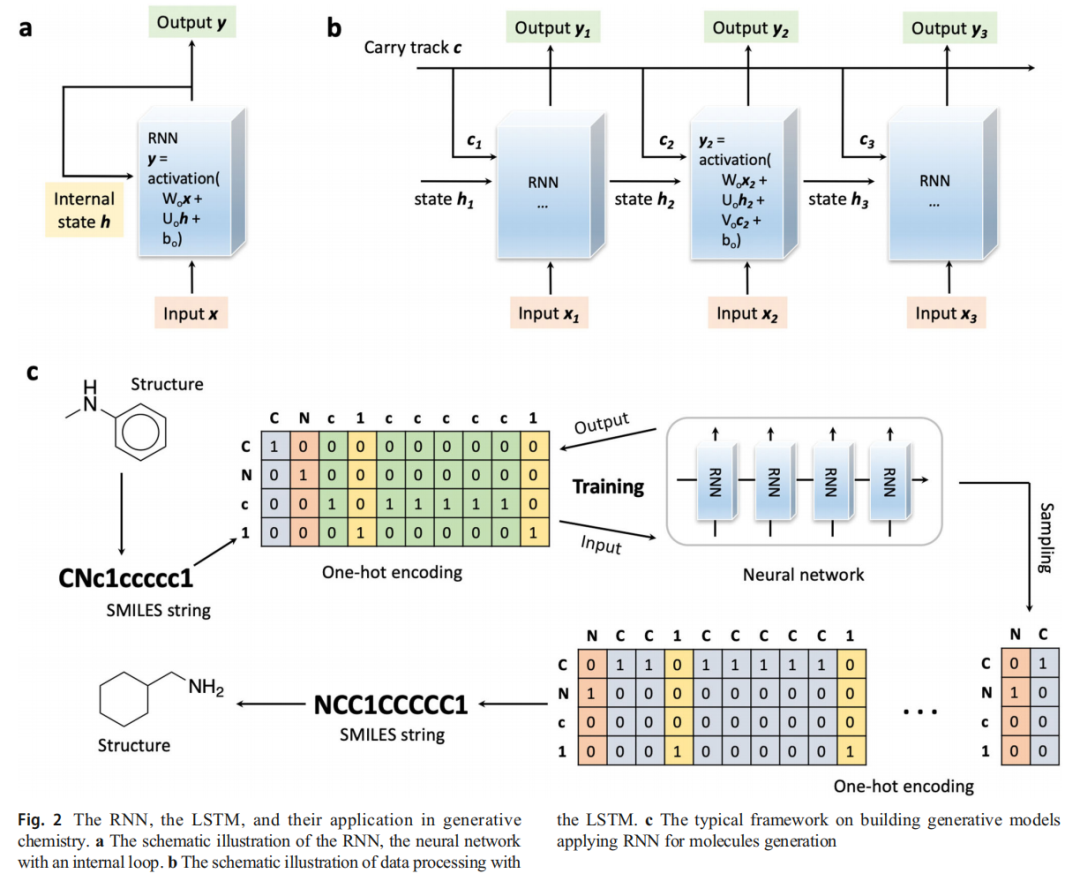
在药物生成中的应用流程
1. 收集训练分子(ChEMBL 等大库)
2. 计算 SMILES 字符串
3. One-Hot 编码(每个字符 → 唯一二进制向量,含起始符 "G" 与终止符 "E")
4. 训练 LSTM:给定前 n 个字符,预测第 n+1 个字符的概率分布
5. 采样生成新 SMILES → 解码为分子图迁移学习策略
两阶段训练是 RNN 在生成化学中的标配:
- 1. 预训练:在数十万至百万量级化合物(如 ChEMBL 全库)上学习通用化学语言
- 2. 微调:在数百至数千个靶标特异性活性分子上精调,使生成分布"聚焦"于目标化学空间
代表性工作
研究团队 | 核心贡献 |
|---|---|
Gupta et al. | LSTM 迁移学习生成 PPARγ 和胰蛋白酶配体库,~90% 分子为新颖独立结构;拓展至片段生长策略 |
Segler et al. | 实现完整"设计—合成—测试"闭环,目标预测模型作为评分函数 |
Merk et al. | 前瞻性研究:合成测试 5 个 AI 设计化合物,4 个对 PPAR 显示纳摩尔至低微摩尔活性 |
Moret et al. | 化学语言模型(CLM),在低数据情境下的早期分子设计 |
Zheng et al. | 基于 GRU 的拟生源分子生成器,拓展生源化学多样性空间 |
3.2 变分自编码器(VAE)
自编码器基础
自编码器(AE)由编码器(高维 → 低维潜空间)与解码器(低维 → 重建)构成,目标是学习分子的压缩表示。然而,标准 AE 将每个分子映射到潜空间中的单一固定点,导致解码器在随机采样时失效。
VAE 的核心创新
VAE(Kingma & Welling, 2013)将分子映射为潜空间中的统计分布参数(均值 μ 与方差 σ),而非固定点:
- • 编码分布:
q_φ(z|x) - • 解码分布:
p_θ(x|z) - • 联合模型:
p(x,z) = p_θ(x|z)·p(z)
训练目标包含两部分损失:
- • 重建损失:评估解码分子与输入的匹配度
- • 正则化损失(KL 散度):防止潜空间过拟合,保证其连续性与可采样性
训练过程的随机性使得同一潜空间点可解码出多样化结构,形成结构化、连续的化学空间表示。
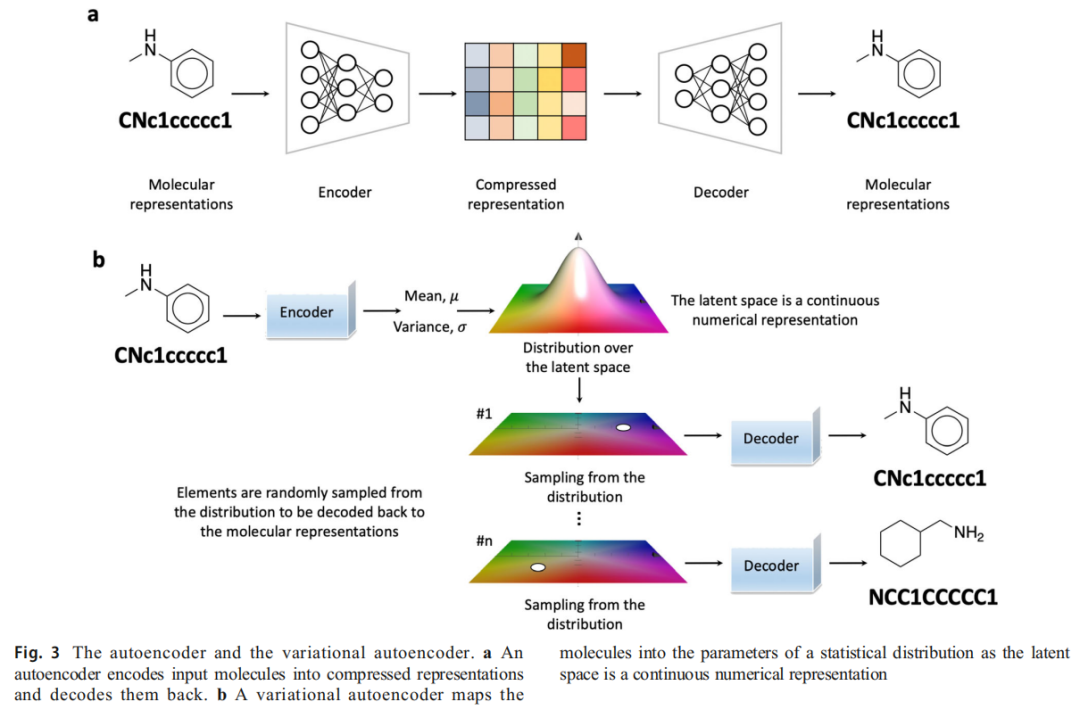
分子编辑能力
在潜空间中,特定轴向对应特定的分子属性变化(称为"概念向量"),通过沿特定向量方向移动,可实现受控的分子性质编辑——这是 VAE 区别于 RNN 的关键优势。
两类输入表示
基于 SMILES 的 VAE
- • Gómez-Bombarelli et al.(2016,首个 VAE 分子设计系统):编码器用三层 CNN + 全连接层,解码器用三层 GRU;联合属性预测 MLP 优化目标性质
- • Blaschke et al.:针对多巴胺受体 2(DRD2),集成 SVM 分类器评估新生成分子
- • Sattarov et al.:VAE + 生成拓扑映射(GTM)辅助采样,聚焦特定性质的化合物库生成
基于分子图的 VAE
- • NeVAE(Samanta et al.):节点表示原子(One-Hot 编码),边权重表示键类型,解决图结构不规则性
- • GraphVAE(Simonovsky et al.):核心假设为概率全连接图,节点/边的存在性为独立随机变量
- • DeLinker(Imrie et al.):将两个片段连接为完整分子,重度依赖三维空间距离与方位信息
3.3 对抗自编码器(AAE)
与 VAE 的关键差异
AAE 在 VAE 的基础上引入判别器(Discriminator),形成三模块结构:编码器、解码器、判别器。
判别器的作用:区分编码器输出的潜分布 q_φ(z) 与预设先验分布 p(z),以对抗代价驱动编码器迫使潜空间服从指定先验。
与 VAE 的 KL 散度约束相比,AAE 的对抗训练机制允许使用非高斯先验,提供更强的分布建模灵活性。
三阶段训练流程
- 1. 编码器 + 解码器联合训练,最小化重建损失
- 2. 判别器训练,准确区分
q_φ(z)与p(z) - 3. 编码器训练,最小化判别器的对抗代价

代表性工作
研究 | 分子表示 | 核心贡献 |
|---|---|---|
Kadurin et al.(druGAN) | MACCS 指纹 | 利用 MCF-7 细胞系数据生成潜在抗癌分子;移除批归一化层以避免生成信号被掩盖 |
Polykovskiy et al. | SMILES(LSTM) | 条件 AAE,针对 JAK3 设计分子,融合生物活性、溶解度、可合成性多约束;经分子对接与 JAK2/3 体外抑制实验验证 |
Shayakhmetov et al. | SMILES | 双向 AAE,将分子结构与基因表达变化联合建模,生成能诱导特定基因表达改变的分子 |
3.4 生成对抗网络(GAN)
卷积神经网络基础
CNN 的卷积层具有两项关键特性:
- • 局部模式学习:在感受野(Kernel)内聚焦局部特征,低层学习边缘/片段,高层抽象复杂模式
- • 位置不变性:学到的特征可在输入任意位置被识别,参数效率高
池化层(Pooling)对特征图降采样,减少参数量;多个滤波器(Filters)并行学习输入的不同方面。
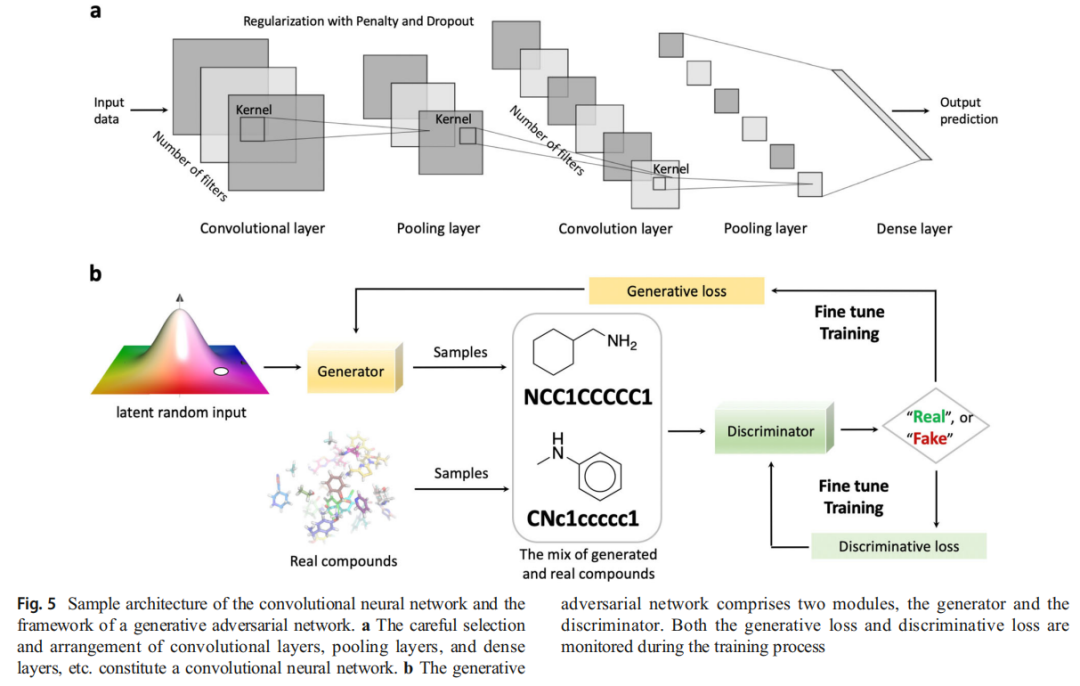
GAN 原理
GAN(Goodfellow, 2014)由**生成器(Generator)与判别器(Discriminator)**对抗训练:
- • 判别器:识别真实数据与生成数据的差异,最大化判别损失
- • 生成器:持续生成逼真样本以欺骗判别器,最小化生成损失
训练目标是达到纳什均衡——判别器无法再区分真实与生成样本。
在分子生成中,生成器输出 SMILES 字符串、分子图或指纹;生成分子与真实化合物混合后输入判别器进行真假鉴别。
GAN 的固有挑战
- • 训练不稳定性:两个损失函数的同步优化容易导致单边梯度主导(判别器或生成器过强)
- • 化学空间覆盖受限:为欺骗判别器,生成器只能在真实化合物定义的化学空间内探索,创新性受限
代表性工作
研究 | 方法亮点 |
|---|---|
Guimaraes et al.(ORGAN) | 目标强化 GAN,在判别器奖励之外引入领域特异性目标(如药物相似性),兼顾分布匹配与性质优化 |
Maziarka et al.(Mol-CycleGAN) | 基于 Junction Tree VAE 潜空间的循环 GAN,生成与输入高度相似但指定性质更优的结构 |
Méndez-Lucio et al. | 连接化合物设计与系统生物学:给定靶标基因表达特征,生成类活性分子;两阶段训练策略 |
Prykhodko et al.(latentGAN) | AE + GAN 融合:heteroencoder 将 SMILES 映射到潜向量,GAN 直接在潜空间操作后回译为分子结构 |
四、生成模型评估方法
4.1 通用评估指标
指标 | 方法 | 特点 |
|---|---|---|
Inception Score (IS) | 计算生成分子与训练集分布的 KL 散度 | 一维评分,无法区分不同失败模式 |
Fréchet Inception Distance (FID) | 比较真实与生成数据在 Inception 模型特定层的潜向量分布(多变量高斯)距离 | 可用于无标签数据 |
Precision & Recall (PRD) | Sajjadi et al. 提出,将分布差异分解为精度(质量)与召回率(覆盖度)两个独立维度 | 解决 IS/FID 的一维局限 |
4.2 化学特异性评估
药物生成中,模型性能评分与真实可用性之间存在鸿沟——高分不等于可合成(Gao et al., 2020)。
可合成性评估方法
- • SA Score(合成可及性评分):Ertl et al. 提出,基于片段贡献与复杂性惩罚估算合成难度,来源于对已合成化学品的历史知识挖掘
- • SCScore:基于反应数据库学习的合成复杂性评分(Coley et al.)
- • 结构过滤器:ChemAxon Structure Checker 等工具过滤化学不合理结构
常见做法:在生成流程末端加入 SA Score 过滤,或将其直接作为强化学习的奖励信号(如 Gómez-Bombarelli et al. 的工作)。
五、技术对比总结
维度 | RNN/LSTM | VAE | AAE | GAN |
|---|---|---|---|---|
分子表示 | SMILES | SMILES / 分子图 | SMILES / 指纹 | SMILES / 分子图 |
生成机制 | 序列逐字符采样 | 潜空间连续采样 | 潜空间 + 对抗分布匹配 | 生成器对抗采样 |
化学空间探索 | 受训练集分布约束 | 连续插值,可控编辑 | 比 VAE 更灵活的先验 | 受真实数据边界限制 |
属性优化 | 强化学习 / 迁移学习 | 联合属性预测 MLP | 条件生成 | 领域特异性奖励 |
训练稳定性 | 高 | 较高 | 中 | 低(对抗博弈) |
可解释性 | 低 | 中(潜空间可视化) | 中 | 低 |
代表数据库 | ChEMBL | ZINC / QM9 | ZINC / MCF-7 | ZINC / LINCS |
六、挑战与未来展望
6.1 三维几何感知不足
现有主流模型多基于 2D SMILES 或拓扑分子图,忽略了手性(Chirality)和构象(Conformation)对受体—配体相互作用的关键影响。基于分子图的工作(NeVAE、GraphVAE、DeLinker)已引入键长、键角信息,但三维感知的系统整合仍是瓶颈。
6.2 大分子设计的匮乏
目前生成化学高度集中于小分子领域,原因在于蛋白质、多肽等大分子数据积累相对不足。随着蛋白质结构数据库持续扩张(AlphaFold 等带来的数据红利),从头蛋白质生成(de novo protein design)有望成为下一个突破口,但需要能够感知折叠与构象的新型分子表示。
6.3 可合成性的系统性忽视
生成模型可产出量化评分优异但实验室无法合成的"纸面分子"。将可合成性约束深度融入生成过程(而非作为后处理滤波器)是迫切的工程需求。
6.4 闭环自动化
理想的 AI 驱动药物发现闭环:
HTS 筛选命中化合物
↓
迁移学习训练生成模型
↓
生成 + ML 分类筛选候选分子
↓
机器人合成 + 生物活性测试
↓
新活性分子回馈模型 → 循环迭代Segler et al. 的工作已概念验证了这一框架,但实现全自动化闭环仍面临合成机器人、高通量检测与 AI 模型三者的系统集成挑战。
写在最后
本综述的核心价值在于提供了一张从基础设施到前沿架构的完整技术地图。对于初学者,它是理解生成化学知识体系的优质入口;对于研究者,各架构的详细对比与案例汇总(Table 4 收录 19 项代表性工作)具有直接参考价值。
值得特别关注:
- 1. 迁移学习的战略地位——在小数据的药物靶标领域,大库预训练 + 靶标微调的两阶段策略几乎是所有生成方法取得成功的必要条件。
- 2. 分子表示的多元化趋势——从 SMILES 到分子图,表示方式的选择深刻影响生成质量与下游任务的适配性。图表示在保留拓扑信息方面的优势正推动 GNN 在生成化学中的地位快速上升。
- 3. 评估方法与实用性的断层——当前学术评估指标(IS、FID、PRD)与真实药物研发需求(可合成性、体内活性)之间存在显著鸿沟,建立面向实用的生成化学评估体系是迫切的社区需求。
本文撰写于 2021 年初,此后领域发展迅猛。扩散模型(Diffusion Model)、大语言模型(LLM)与分子基础模型(如 ESMFold、MolBERT)的兴起,正在重塑生成化学的技术格局——这些是本综述未能涵盖但值得密切追踪的新方向。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-18,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号