AI 蛋白设计,到底该信哪个分数?

AI 蛋白设计,到底该信哪个分数?

DrugIntel
发布于 2026-06-03 19:04:13
发布于 2026-06-03 19:04:13
3,766 个实验结合子、15 个靶点的大规模 meta 分析,系统回答了「计算 筛选该用哪个指标」这个长期没有标准答案的问题。

作者 Max D. Overath†, Andreas S. H. Rygaard†, Christian P. Jacobsen, Valentas Brasas, Oliver Morell, Pietro Sormanni, Timothy P. Jenkins 单位 丹麦技术大学(DTU)生物技术与生物医学系;剑桥大学 Yusuf Hamied 化学系 类型 bioRxiv 预印本(2025 年 9 月,未经同行评审) DOI10.1101/2025.08.14.670059 代码链接:https://github.com/DigBioLab/de_novo_binder_scoring
速览
设计 de novo 蛋白结合子已越来越容易,但在合成与实验验证之前如何排序候选,长期依赖经验性启发式(heuristics),缺乏系统比较。本文做了三件事:
- 1. 汇总了来自多项已发表研究的 3,766 个带实验结合数据的 de novo 结合子,覆盖 15 个结构与功能差异极大的靶点;
- 2. 用四种结构预测工具重折叠每一个复合物(AF2 initial guess、ColabFold、AlphaFold3、Boltz-1),为每个设计提取 200+ 个结构、能量、置信度与序列特征;
- 3. 系统评估了哪些指标(及组合)最能预测实验结合成功。
核心结论:AlphaFold3 的 ipSAE_min 是跨靶点表现最稳、最具区分力的单一指标,其平均精度(AP)约为常用 ipAE 的 1.4 倍;再叠加正交的物理化学界面描述符(如 Rosetta ΔG/ΔSASA、界面形状互补性)可进一步提升;但简单线性模型用好少数几个特征即可,堆特征、加交互项或换用 XGBoost 均无额外收益。作者同时给出了可直接落地的阈值与筛选流程,并开源了完整数据与代码。
关键数字 | 值 |
|---|---|
汇总结合子总数 | 3,766 |
靶点数 | 15 |
真实结合子(阳性) | 436(11.6%) |
每个设计的特征数 | 200+ |
结构预测工具 | 4 |
靶点长度范围(残基) | 60–621(均值 174,中位 101) |
流水线提速 | 每个设计 1802 s → 230 s(↓87%) |
1. 背景:为什么「筛选」才是真正的瓶颈
近两年,RFdiffusion、BindCraft、AlphaProteo 等方法已经可以只凭靶点结构,无需天然模板,直接生成高亲和力结合子,并被广泛用于治疗、诊断与基础研究。
但真正的成本在后端:设计软件往往一次产出成百上千个候选,而能在体外实测中结合的常常只有个位数。逐一合成、做 BLI 或细胞分选筛选既昂贵又耗时。因此,in silico 预筛成为整个 pipeline 的核心难点——而且这是一个特别棘手的筛选问题,因为候选池往往是一群高度相似的结构,需要在其中分出高下。
一个关键转折是 Bennett et al.(2023)等工作发现:基于深度学习的结构预测(尤其 AlphaFold2)能在实验前有效排序候选,显著提高成功率。由此衍生的置信度指标——pLDDT、ipAE、ipTM——被证明能预测体外结合,且优于传统的 Rosetta 物理能量。
然而,现状仍有三个明显缺口:
- 没有公认标准:设计成功率高度不稳定,缺乏跨研究通用的候选优先级判定准则;
- 新模型未被系统评测:AF2 之后涌现的 AlphaFold3、Boltz-1、Boltz-2、Chai-1 等,在「预测体外结合」这一任务上谁更强,尚无大规模比较;
- 泛化性存疑:层出不穷的新指标,能否跨大规模、多样化数据集泛化,并不清楚。这一点又被现实加剧——多数 de novo campaign 只产出极少量验证过的结合子,且往往聚焦于相关靶点,使得难以在规模上 benchmark。
本文正是针对这些缺口而来。
2. 数据集:3,766 个结合子 / 15 个靶点
作者从多篇已发表研究中汇总数据(见下表),构建了一个跨靶点类别、跨设计协议的大规模数据集。靶点涵盖:
- 受体酪氨酸激酶(RTK):EGFR、LTK、FGFR2、胰岛素受体(InsulinR)、TrkA
- 病原体来源抗原:SARS-CoV-2 RBD、VirB8
- 细胞因子受体 / 免疫调节因子:IL2Rα、IL7Rα、IL10Rα、PD-L1
- 其他:原癌蛋白 MDM2、蛇毒短链 α-神经毒素(sntx)、两个肽-MHC 复合物(pMHC)
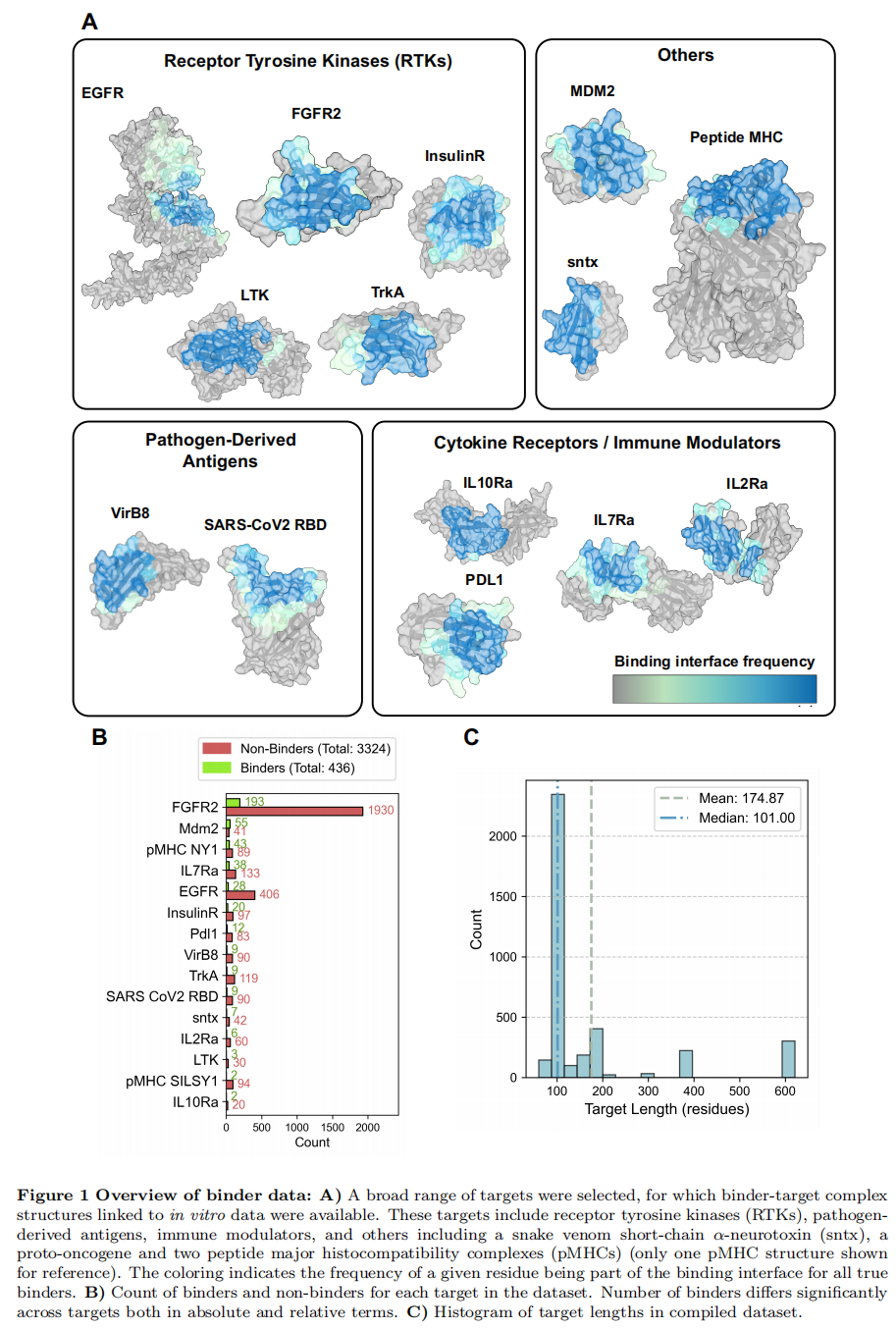
将真实结合子的相互作用残基映射回靶点结构后可见,结合位点高度保守(Fig. 1A)。
2.1 数据来源(节选汇总)
数据来源 | 代表靶点 | 结合判定方式 |
|---|---|---|
Adaptyv Bio R1 & R2 (2025) | EGFR | BLI 测得 K_D < 10,000 nM |
Bennett et al. (2023) | LTK、IL10Rα、IL2Rα | 细胞分选 Sc50 < 4000 nM,部分 BLI 验证 |
Cao et al. (2022) | VirB8、InsulinR、TrkA、FGFR2、EGFR、IL7Rα、SARS-CoV-2 RBS | Sc50 < 4000 nM |
Watson et al. (2023, RFdiffusion) | PD-L1、IL7Rα、TrkA、InsulinR、MDM2 | BLI 响应 > 阳性对照 50%(10,000 nM) |
Johansen et al. (2025) | 两个 pMHC(NY-ESO / SILSY) | 分选前后 log2FC,无单一阈值,部分 BLI |
Torres et al. (2025) | sntx | 酵母展示分选,部分 BLI |
2.2 数据集的两个关键特征(也是后续分析的难点)
- • 极度不平衡:仅 436 个(11.6%)为真实结合子,且各靶点的测试数与阳性比例差异极大(Fig. 1B)。这直接决定了后文用平均精度(AP)而非 AUROC 作为主评价指标。
- • 标签噪声不可忽视:结合判定未跨研究标准化——亲和力阈值与检测形式(BLI、酵母/细胞分选 Sc50、log2FC 等)各不相同(见 Table 1)。
为排除数据冗余带来的偏倚,作者对靶点与结合子序列相似性都做了评估(Fig. S1):除两个 pMHC 靶点之间、以及 pMHC NY1 结合子内部存在预期内的相似性外,整体序列多样性都很高——结论不是被冗余撑起来的。此外,对 Bennett 与 Cao 的数据按非结合子∶结合子 = 10∶1 进行了下采样,Adaptyv 数据中的抗体类结合子被剔除,以保持「纯 de novo」聚焦。
3. 打分流水线与提速
3.1 流水线设计(Fig. 2A)
输入为各设计工具产出的结合子–靶点复合物 PDB(binder = 链 A,target = 链 B),并配一个 CSV 指定靶点子链范围与 MSA 设置。不同工具的处理方式不同:
- • AF2 initial guess:用输入 PDB 固定靶点结构,仅重预测结合子(为高通量而生,作为本研究的参照基线);
- • ColabFold / Boltz-1 / AF3:提取结合子序列 + 相关靶点子链,重预测整个复合物;每条唯一靶点链用 MMseqs2 生成一次 MSA 并复用,再转成各模型所需格式。
随后从各模型输出中抽取置信度分数,并统一计算 ipSAE、pDockQ、pDockQ2、ipAE、LIS(AF2 initial guess 除外),再对最高置信复合物计算一系列结构指标及模型间两两 RMSD——合计 200+ 个特征/设计。
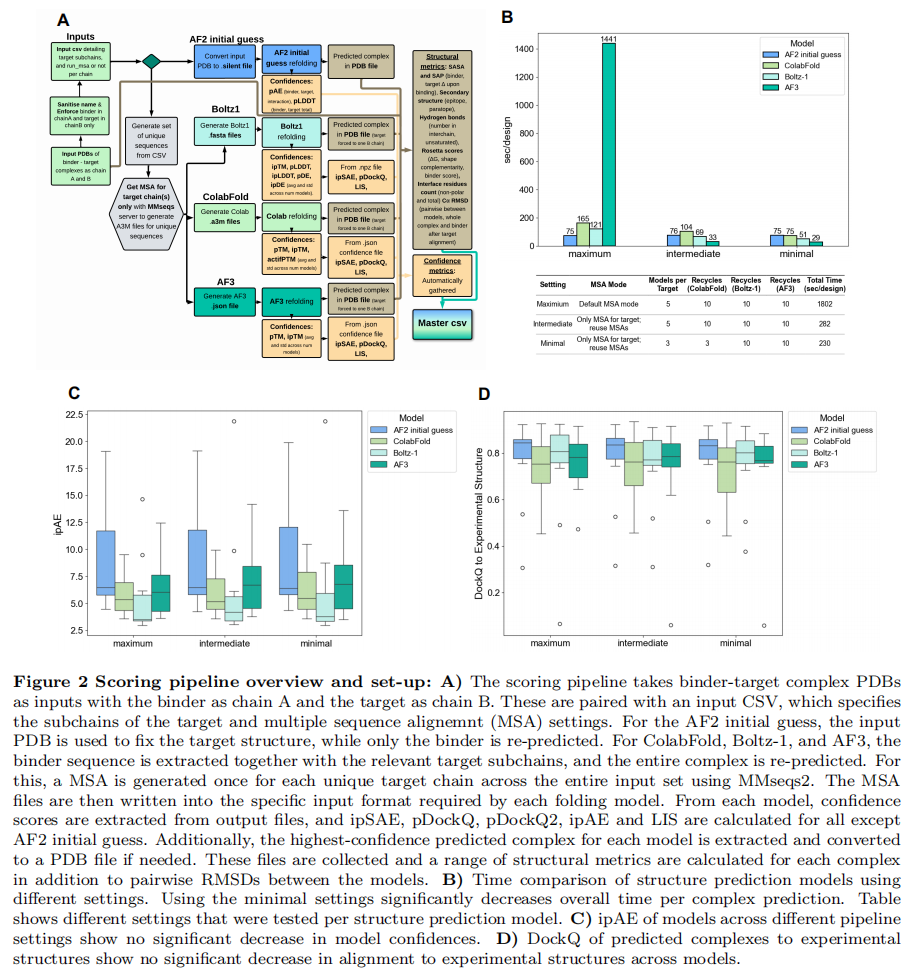
3.2 三档配置与提速结果(Fig. 2B–D)
为压缩「结构预测耗时」这一主要瓶颈,作者在 56 个真实结合子(覆盖 15 个靶点 + 3 个额外靶点;其中 12 个有实验解析结构)上测试了三档配置:
配置 | MSA 策略 | 模型数 | 每个设计总耗时 | AF3 耗时 |
|---|---|---|---|---|
Maximum | 各工具默认成对 MSA | 5 | 1802 s | 1441 s |
Intermediate | 仅靶点单一 MSA,复用 | 5 | 282 s(↓84%) | 33 s(↓97%) |
Minimal(最终采用) | 仅靶点单一 MSA + 减 recycle | 3 | 230 s(↓87%) | 29 s(↓98%) |
AF3 的提速最为戏剧化,主因是跳过了基于 JackHMMER 的 MSA 生成。关键是:用 12 个实验结构验证发现,ipAE(模型质量)与 DockQ(结构吻合度)在三档之间没有显著下降——即提速几乎不以精度为代价。因此后续分析全部采用 Minimal 配置(运行于单张 NVIDIA L40S)。
4. 核心指标:ipSAE 是什么,为什么 _min 最强
要理解本文的核心结论,需要先厘清几个置信度指标的差异。
4.1 ipSAE 的设计动机与公式
ipSAE(interaction prediction Score from Aligned Errors)由 Dunbrack(2025)提出,可视为对 ipTM 的「界面聚焦式」修正:
- • 它与
ipTM算法相似,但只纳入 pAE < cutoff(默认 <10)的链间残基对; - • 它按界面规模动态调整
d0(d0随界面残基数的平方根增大)。
其计算形式为(A→B 方向):
ipSAE(A→B) = max over i∈A [ mean over j∈B, PAE_ij < cutoff ( 1 / (1 + (PAE_ij / d0)^2) ) ]直观含义:只统计真正自信的界面接触,并对「小而自信」的界面打折——因为很短的界面在物理上不太可能形成真实结合。这使得 ipSAE 比 ipAE 更具一致性、更不依赖具体靶点。
4.2 _min 的由来
由于 A→B 与 B→A 不对称,作者保存了两个方向的最大与最小值:
- •
ipSAE_max:标准实现,取较大值; - •
ipSAE_min:取较小值——「最弱的一环」最能反映结合状态。
作者还探索了在 A 维度上把 max 算子替换为 avg / min 的变体(ipSAE_avg、ipSAE_min_in_calculation),以及若靶点有多个子链则在「有实际相互作用残基」的方向上取均值。
4.3 核心指标速查表
指标 | 含义 | 计算于哪些模型 |
|---|---|---|
pLDDT | 每残基置信度 | 全部 |
ipAE(pae_interaction) | 结合子链到其他链的平均链间 pAE | 全部 |
ipTM | 界面预测 TM-score(链相对位置置信度) | 全部 |
ipSAE / ipSAE_min | ipTM 的界面聚焦修正,pAE<10,d0 动态;_min 取双向最小 | AF3 / ColabFold / Boltz-1 |
LIS | 链间接触(pAE<12)反转后取均值(0–1) | AF3 / ColabFold / Boltz-1 |
pDockQ / pDockQ2 | 由 pLDDT + 界面预测的 DockQ | AF3 / ColabFold / Boltz-1 |
DockQ | 与参考结构的界面相似度(此处以输入结构为参考) | 全部 |
interface_ΔG | Rosetta 估计的界面结合能 | 输入 + 各模型 |
interface_ΔG/ΔSASA | 结合能按界面大小归一(惯例 ×100) | 输入 + 各模型 |
interface_sc | 界面形状互补性 | 输入 + 各模型 |
ΔSAP(sap_delta) | 空间聚集倾向差值,疏水性代理 | 输入 + 各模型 |
RMSD_binder(input vs AF3) | 对齐靶点后,输入与 AF3 预测结合子的 Cα RMSD | 跨模型对比 |
4.4 四个工具的角色对比
工具 | 版本 | 处理方式 | 备注 |
|---|---|---|---|
AF2 initial guess | dl_binder_design v1.0.0 | 固定靶点,仅重预测结合子 | 高通量参照基线;不计算 ipSAE/LIS |
ColabFold | localcolabfold v1.5.5(AF-multimer) | 重预测整个复合物 | MMseqs2 MSA |
AlphaFold3 | v3.0.1 | 重预测整个复合物 | 综合最佳 |
Boltz-1 | Boltz-1x v1.0.0 | 重预测整个复合物 | 开源 AF3 类模型,最接近 AF3 但仍略逊 |
5. 哪些特征最能预测结合
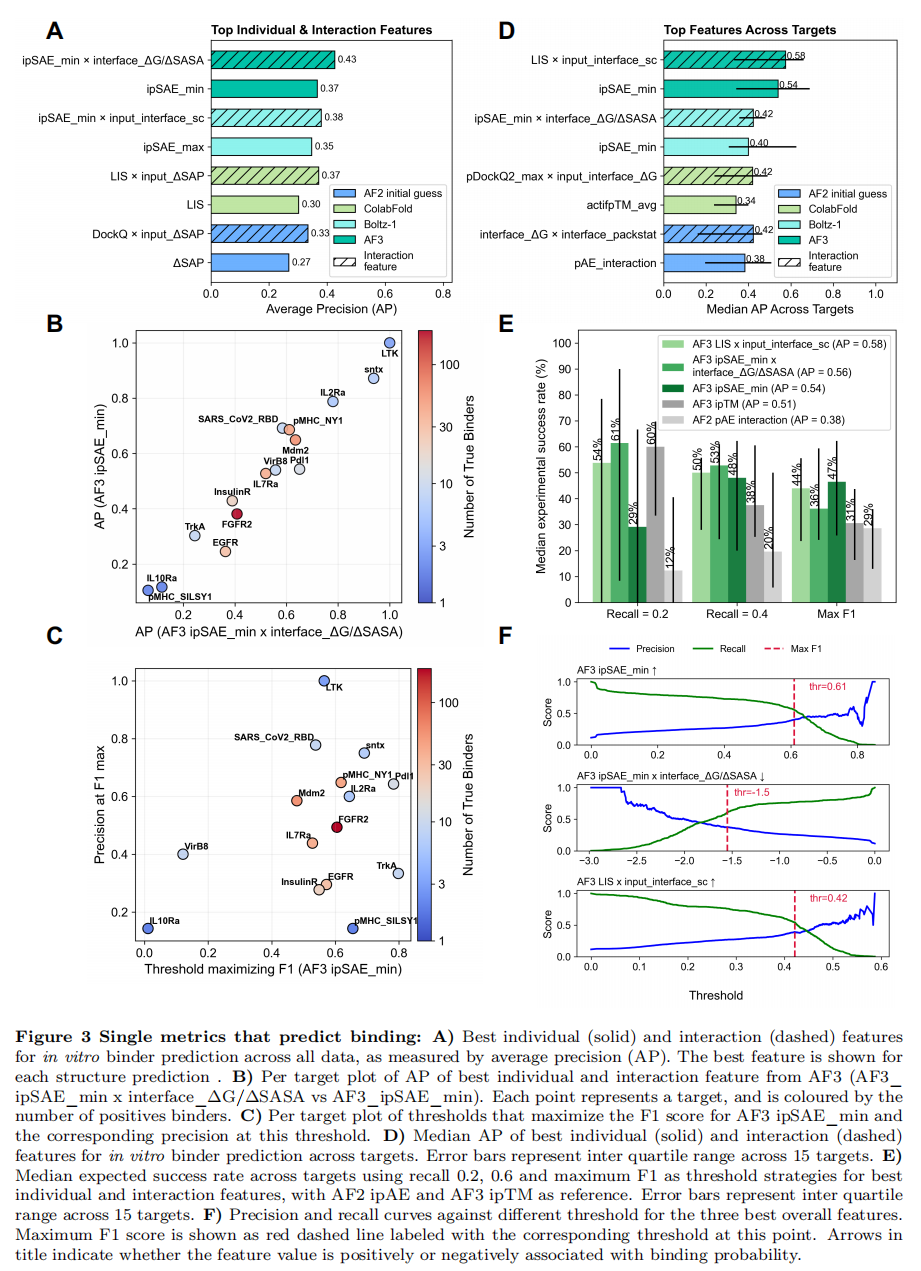
5.1 单特征:全数据下的最佳是 AF3 ipSAE_min
用 AP 衡量各特征区分结合/非结合的能力,各模型的最佳单特征为:
- • AF3 / ColabFold / Boltz-1:
ipSAE_min、ipSAE_max、LIS——全部是基于 pAE 矩阵、且仅捕获高置信界面的分数; - • AF2 initial guess:
ΔSAP。
全数据下的最佳单一指标是 AF3 ipSAE_min。 作者也提醒:部分数据此前已用 AF2 initial guess 的 ipAE/pLDDT 预筛过(Fig. S9、S10),可能人为压低了这两个指标在本数据上的表观预测力。
5.2 交互特征:置信度 × 物理化学描述符
作者进一步引入交互项(两特征的乘积 f_i × f_j)。结果显示,加入交互项后各模型预测力都一致提升,最佳组合为:
模型 | 最佳交互组合 |
|---|---|
AF3 | ipSAE_min × interface_ΔG/ΔSASA |
ColabFold | ipSAE_min × input_interface_shape_complementarity |
Boltz-1 | LIS × input_ΔSAP |
AF2 initial guess | DockQ × input_ΔSAP |
规律很清晰:除 AF2 initial guess 外,最优组合都是「界面聚焦的置信度分数 + 物理化学界面描述符」——说明二者捕获的是正交、互补的信息。整体而言,AF3 在单特征与交互特征上都全面胜出,提示它给出的置信度与复合物结构最准确。
5.3 跨靶点稳健性:换个视角看「中位 AP」
由于数据按靶点不平衡,作者改用跨 15 个靶点的中位 AP(削弱个别靶点的离群影响)重排。各模型 top 单特征:ipSAE_min(AF3、Boltz-1)、actifpTM(ColabFold)、pAE_interaction(AF2)。其中 AF3 ipSAE_min 仍最突出;叠加物理化学描述符(AF3 加形状互补性、Boltz-1 加 ΔG/ΔSASA 等)同样能稳定提升 AP。
一句话:无论看「全数据 AP」还是「跨靶点中位 AP」,AF3
ipSAE_min都是最佳单一指标;而最强的单一组合基线是 AF3LIS × input_interface_shape_complementarity(中位 AP ≈ 0.58)。
6. 靶点依赖性:为什么有的靶点天生难预测
这是本文最有「降温」意味、也最诚实的一节。
- • 跨靶点波动极大:AF3
ipSAE_min(及其与ΔG/ΔSASA的组合)在不同靶点上的 AP 从 0.1 一路到 1.0;真结合子很少的靶点常是离群点,多半反映统计波动(Fig. 3B)。 - • 阈值稳定,但精度不稳定:让
ipSAE_min的 F1 最大化所对应的阈值,在多数靶点落在 0.5–0.8;但对应的精度仍在 0.1–1.0 间剧烈波动(Fig. 3C)。各靶点的 top 特征也相当不同(Fig. S11)。 - • 什么样的靶点更好预测?(Fig. S12)
- •
ΔSAP(疏水性)与 AP 的相关性仅在 AF2ipAE上成立(Pearson r ≈ −0.68,p = 0.005); - • 但在 AF3
ipSAE_min上不成立(r ≈ −0.04,p = 0.89)——说明ipSAE_min不依赖疏水埋藏,捕获的是不同维度的界面质量; - • AF3
ipSAE_min的 AP 反而与界面氢键比例正相关、与ΔG/ΔSASA负相关——即「界面能量越密集的靶点越好预测」。但这些相关都偏弱、未达显著,且主要由少数靶点驱动。
- •
7. 阈值策略:回顾性分析 + 交叉验证
候选筛选最终要落到「卡哪个阈值」。作者用留一靶点交叉验证(每次留出一个靶点,阈值在其余靶点上选定),对最佳单特征与交互特征做了回顾性评估,并以常用的 AF2 ipAE 与 AF3 ipTM 作基线。三种实用阈值策略对应:recall = 0.2 / recall = 0.4 / 最大化 F1(Fig. 3E)。
结论:
- • AF3
ipSAE_min及交互特征AF3 ipSAE_min × interface_ΔG/ΔSASA、AF3 LIS × input_interface_shape_complementarity在所有阈值下都优于 AF2ipAE; - • AF3
ipTM在 recall 0.4 与 max F1 时被超越,但在 recall 0.2 时排第二,且 IQR 更小——说明ipTM在「宁缺毋滥」的低召回区依然稳健。
精度-召回曲线(Fig. 3F)给出的 F1 最大化阈值:
- •
AF3 ipSAE_min:0.61 - •
AF3 ipSAE_min × interface_ΔG/ΔSASA:−1.5(越小越可能结合) - •
AF3 LIS × input_interface_shape_complementarity:0.42
此外,对 ipSAE_min 做的「逐步增加训练靶点」分析(Fig. S13)表明:阈值很快稳定,精度与 F1 随训练靶点数增加而稳步提升。
8. 线性模型与贪婪特征选择:多特征真的更好吗?
作者进一步检验:把多个特征线性组合(而非乘积),能否跨靶点提升表现?方法是逻辑回归 + 贪婪前向特征选择 + 嵌套留一组交叉验证(15 个靶点逐一留出):
- • 所有数值特征 z-score 标准化;类别不平衡用
class_weight="balanced"处理; - • 内层用 l1 惩罚逻辑回归(
liblinear),以中位 AP 选特征,AP 增益 < 0.005 即早停,防过拟合; - • 分别对每个工具的「单特征」「单特征 + top50 交互项」以及「跨模型合并」做了实验。
结果(Fig. 4A,Fig. S14–S15):
- • 没有任何模型超过最强基线
LIS × input_interface_shape_complementarity(中位 AP = 0.58,IQR 0.33–0.66); - • 但用 AF3 单特征训练的模型中位 AP = 0.57(IQR 0.40–0.66,方差更小),超过了最佳单特征
ipSAE_min(≈0.54); - • 内层平均只选了 2–5 个特征就停止改进;
- • 跨模型合并特征 → 不提升(像是引入噪声);加交互项 → 不提升(选入更少,说明多数交互项不跨靶点泛化);换 XGBoost → 仍不提升(Fig. S15)。
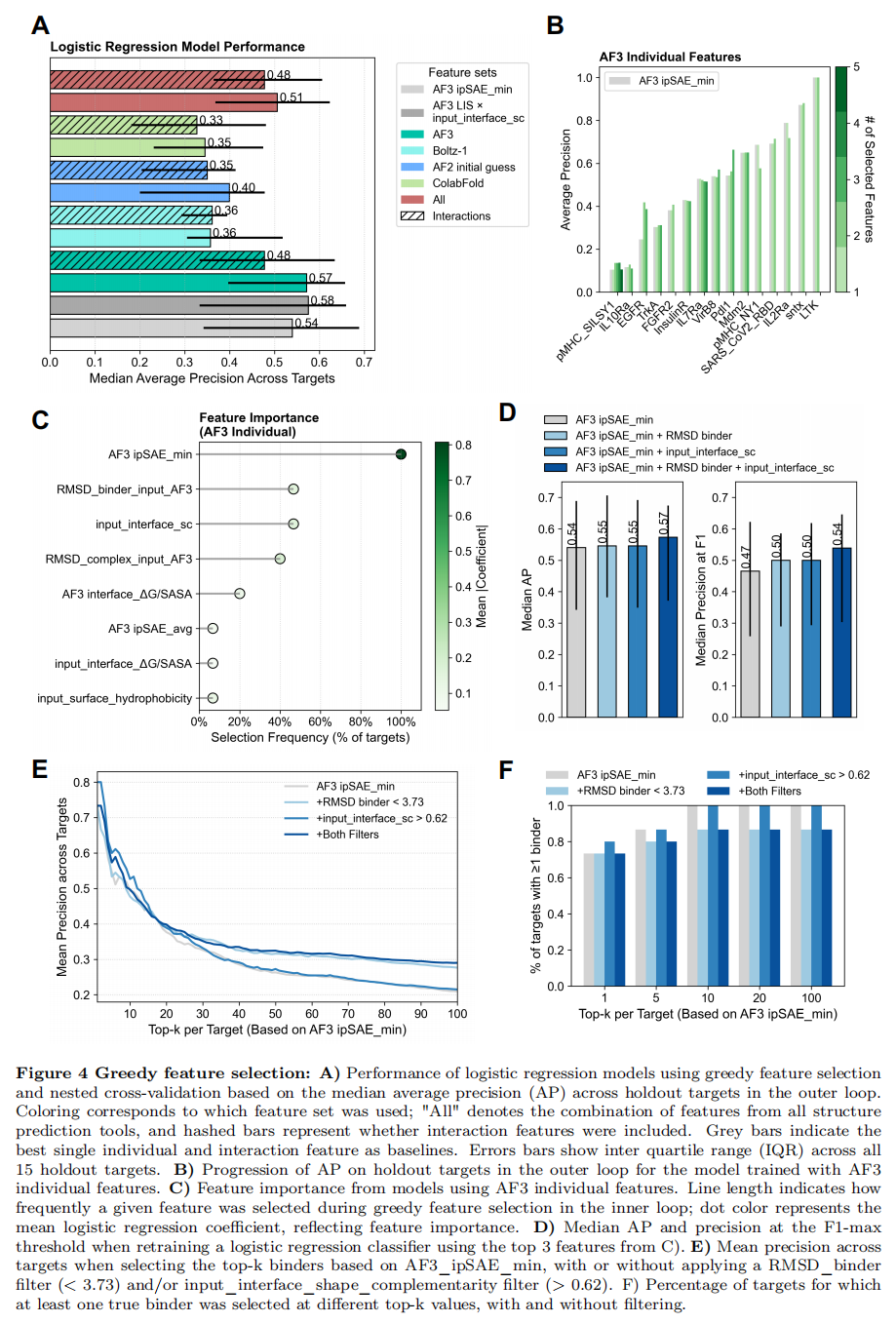
特征重要性(AF3 单特征模型,Fig. 4C):ipSAE_min在所有折中均被选中,稳健性再次得到印证;之后选择频率与系数断崖式下降。值得注意的是,两个结构对比类特征频繁入选——RMSD_binder(对齐靶点后输入 vs AF3)与 RMSD_complex(整复合物 输入 vs AF3);此外 input_interface_shape_complementarity 与 interface_ΔG/ΔSASA 也常被选中。
**三特征模型(Fig. 4D):**取 top-3 特征 AF3 ipSAE_min + RMSD_binder + input_interface_shape_complementarity 重训逻辑回归,同时加这两个特征比只加其中之一带来的 AP / 精度 / F1 提升都更大——即少量补充特征确实能进一步抬升实验成功率。
方法学结论:跨多样化靶点真正稳定有用的特征屈指可数;简单线性模型用好这几个,反而最可靠。这也凸显了「为全新靶点构建可泛化预测模型」的本质困难。
9. 落地建议:可直接复用的筛选 SOP
作者把上述发现凝练成两套可直接执行的筛选策略(任选其一或组合):
方案 A — 单刀直入,卡一个阈值(三选一)
AF3 ipSAE_min > 0.61
AF3 ipSAE_min × interface_ΔG/ΔSASA < -1.5 # 越小越可能结合
AF3 LIS × input_interface_shape_complementarity > 0.42方案 B — 先粗筛,再排序(推荐用于大批量)
Step 1 预筛选(作用于「设计的输入结构」,无需折叠即可执行):
input_interface_shape_complementarity > 0.62
RMSD_binder < 3.73
Step 2 对通过者按 AF3 ipSAE_min 取 Top-K几条值得记住的实操要点(Fig. 4E–F)
- • 形状互补性这个滤镜很值:它作用在输入结构上,无需等复合物折叠完即可应用,在小 K(约 1–20)时尤其能提升平均精度;
- • 仅用
ipSAE_min(或再加形状互补性),每个靶点只取 10 个候选,就能为全部 15 个靶点各召回至少 1 个真结合子; - •
RMSD_binder滤镜需谨慎:它有时过于严格——在两个靶点上、所有 K 值下都一个都没召回。它能增强预测力,但可能误杀。
10. 局限性与展望
作者对局限性相当坦诚:
- • 数据稀疏:多数靶点的测试设计有限、真结合子常为个位数,限制了模型泛化;
- • 标签噪声:各研究的检测方法与「结合」定义异质,引入噪声;
- • 亲和力缺失:亲和力数据仅小部分可得,未纳入分析。
展望与呼吁:
- • AF3 持续领先,与既往「AF3 在 PPI 预测上更优」的结论一致;但 Boltz-2、Chai-1 等更新模型值得继续评估;
- • 该领域真正需要的,是更标准化、公开的「结构 ↔ 亲和力」数据集,以提升预测力并深化对蛋白–蛋白相互作用的理解。
写在最后
如果说过去两年 de novo 设计解决了「怎么造」,那么「造出来之后该信谁、该挑谁」一直缺一个有规模支撑的答案。这篇覆盖 3,766 个样本的 meta 分析,第一次在足够大的尺度上把这件事讲清楚,并给出简单、可解释、可复现的落地方案:
先用 AF3 重折叠复合物,盯住
ipSAE_min排序,再用界面形状互补性帮你把关。
对任何还在为「挑哪个 binder」头疼的实验室,这都是一份兼具方法学深度与工程实用性的参考。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号