bioRxiv|16万条多肽序列、15个数据集:PPB为多肽AI建立新基准

bioRxiv|16万条多肽序列、15个数据集:PPB为多肽AI建立新基准

MindDance
发布于 2026-06-01 18:17:08
发布于 2026-06-01 18:17:08

BioRxiv. 2026. A Systematic Benchmark for Peptide Property Prediction
GLP-1类多肽药物彻底点燃了生物医药行业的投资与研发热情。大模型在通用场景里大杀四方,但在多肽药物发现这个典型的科学计算场景中,AI的真实底细究竟如何。面对每天都在涌现的新模型、新算法,行业急需一把统一、客观且足够硬核的标尺。近期,西安电子科技大学等团队,在biorxiv发表了一项系统性基准评估工作——PPB(Peptide Property Benchmark)。他们没有去卷更复杂的模型结构,而是把注意力放在了多肽数据表征与评测体系的底层逻辑上,搭建了多肽性质预测基准平台。这项工作不仅给爆火的多肽预测AI卸了妆,更顺手揭示了一个被整个行业长期忽视的聚类瓶颈,为下一代AI制药工具的研发指明了物理与结构交融的新方向。

降糖药神话背后的隐忧:为什么多肽AI预测总在实战中翻车
多肽作为介于小分子和蛋白质之间的生命桥梁,凭借特异性强、毒性低以及相对低廉的合成成本,正在成为新一代疗法的核心支柱。从抗感染、免疫调节到风靡全球的减重降糖,多肽药物的商业价值与临床潜力已被反复验证。然而,传统的多肽开发极度依赖高成本、低通量的实验筛选,这让计算驱动的虚拟筛选与从头设计顺理成章地成为了刚需。
遗憾的是,多肽性质预测领域长期存在两个难以根治的顽疾,导致许多在论文中表现惊艳的AI模型,一到实际湿实验筛选中就大面积失效。
首当其冲的痛点是数据异质性。公开的多肽数据集往往拼凑自不同的文献与数据库,实验背景、筛选标准甚至负样本的构建策略都大相径庭。比如在预测多肽的溶血活性或抗菌活性时,如果负样本只是简单地用随机序列或不相关的胞质蛋白序列填充,模型在训练时学到的可能只是如何区分多肽的定位或合成来源,而不是真正决定生物活性的理化机制。这种带着面具的数据集,训练出来的模型自然无法应对真实的药物理化性质预测。
另一个更具欺骗性的顽疾,则是评估标准的自我美化。在序列建模任务中,很多研究为了图省事,直接采用随机划分来构建训练集和测试集。然而,多肽数据库中存在大量高度相似的序列。如果测试集里混入了大量训练集的近亲,模型即使只靠简单的记忆也能刷出极高的准确率。这种虚高的指标严重高估了模型的泛化能力,一旦面对自然界或人工设计中从未出现过的全新多肽序列,模型就会立刻现出原形。
16万条序列大扫除:多肽AI界迎来硬核大阅兵
为了打破这种各说各话的混乱局面,西安电子科技大学高琳与李鹏勇团队搭建了PPB多肽性质预测基准。他们首先在数据层面进行了一场彻底的大扫除,通过统一的生物学过滤标准,对公开的多肽数据进行了严格的去重、清洗和标准化。
最终,团队整理出了一个包含15个高质量数据集、总计161,571条唯一多肽序列的超大评测空间。这些数据集不仅规模庞大,而且在理化特征、功能活性上实现了极广的覆盖,具体包含12个分类任务与3个回归任务。从抗菌肽(AMP,47,511条)、自组装肽(ASSEM,41,703条)这样的万级大矿,到血脑屏障穿透肽(BBB,521条)、细胞穿透肽(CPP,924条)这样的稀缺小矿,数据集之间的规模差异跨越了两个数量级。序列长度的分布也极为多样,短至2-3个残基的寡肽,长至接近200个残基的大肽,几乎覆盖了多肽药物发现中所有可能遇到的序列空间。
从下图的数据集统计特征中可以看出,不同数据集在序列长度、等电点分布和类别比例上呈现出高度的异质性,这恰恰是真实多肽药物发现场景的忠实映射。
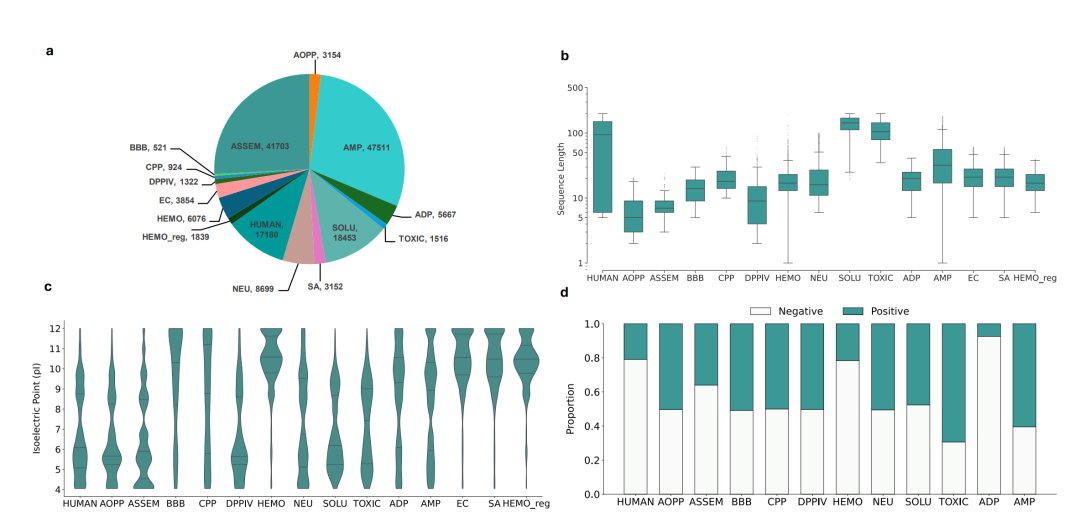
PPB基准中15个数据集的规模、序列长度、等电点与类别比例分布
PPB基准中15个数据集的规模、序列长度、等电点与类别比例分布
为了全面测试不同AI技术路线的战斗力,团队组织了一场覆盖三大主流流派、7种代表性模型架构的硬核大阅兵。
技术流派 | 代表模型 | 特征编码方案 | 核心技术特征 |
|---|---|---|---|
传统机器学习 | XGBoost, Random Forest, SVM | 氨基酸组成频率, 理化描述符, One-hot编码 | 依赖人工构建的物理化学先验特征,解释性强,计算资源消耗极低 |
经典深度学习 | LSTM, Transformer | 端到端序列编码 | 自动学习氨基酸序列的上下文关联,适合中等规模数据集 |
预训练蛋白质语言模型 | ESM-2, PepBERT | 预训练模型冷冻表征 | 在百亿级通用蛋白质序列上进行无监督预训练,提取高维生物学语义 |
在模型训练与评估的流程上,团队同样执行了极其严苛的约束。所有的分类任务均以F1-score作为核心评价指标,以排除数据类别不平衡带来的干扰;回归任务则以 RMSE 作为主标尺。为了测试模型在面对未知序列空间时的真实泛化能力,团队还特别引入了基于序列相似性的划分策略,试图用生物信息学经典的聚类工具MMseqs2将相似多肽打包隔离,从而模拟真实的药物发现场景。
从下图可以直观地看出整个基准评测框架的系统化设计流程,从多任务定义、多源数据清洗、多流派模型构建到双重划分策略下的极限压测,每一步都力求做到严谨、透明且可复现。
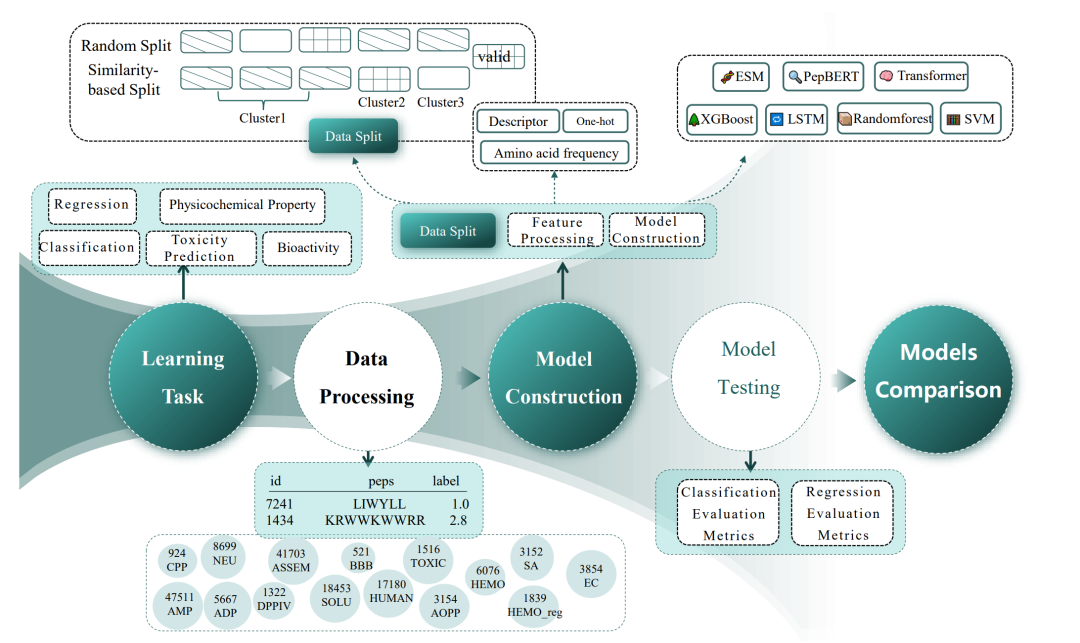
PPB多肽性质预测基准的系统设计与评测工作流
PPB多肽性质预测基准的系统设计与评测工作流
卸妆后的真相:蛋白质大模型降维打击,但多肽聚类其实是个伪命题
这场硬核大阅兵测出了许多让人耳目一新的行业真相,也彻底颠覆了一些流传已久的常识。
以ESM-2为代表的预训练蛋白质语言模型展现出了无可争议的降维打击实力。在12个分类数据集的平均F1-score和3个回归数据集的平均RMSE上,ESM-2均以显著优势占据榜首。在回归任务中,ESM-2的RMSE相较于表现第二好的Transformer模型,在多个数据集上实现了超过10%的误差缩减。
从下图的模型性能对比中可以直观地看到,预训练语言模型在分类和回归任务上的全面领先态势。
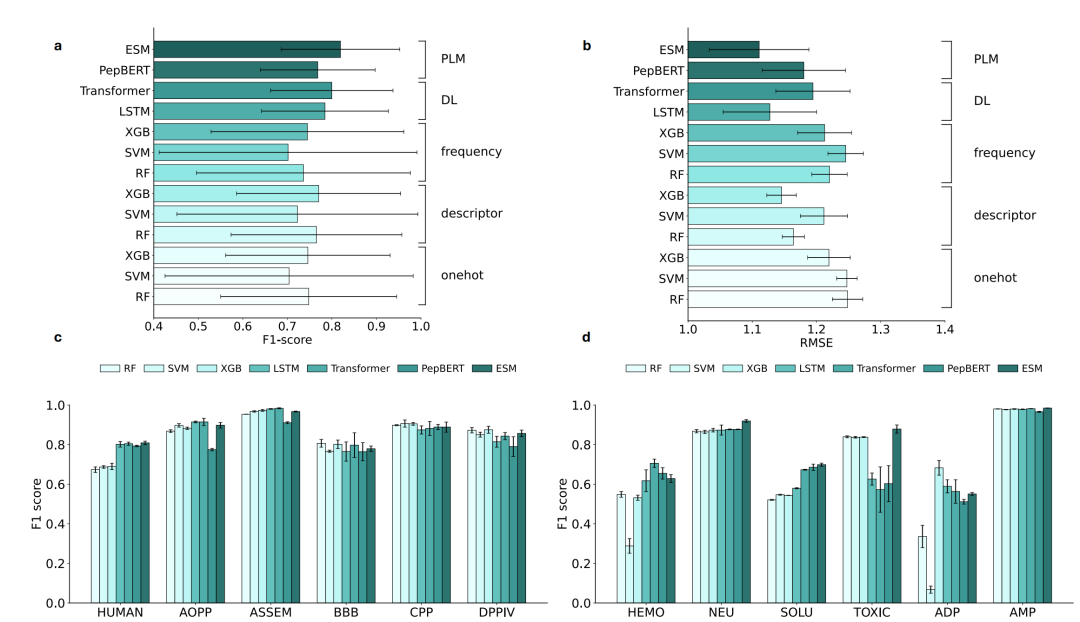
七种模型架构在分类(F1-score)和回归(RMSE)任务上的性能对比
七种模型架构在分类(F1-score)和回归(RMSE)任务上的性能对比
更深层的洞察在于,大模型的优势不仅体现在绝对精度上,更体现在其面对小样本和复杂任务时的惊人稳定性。在血脑屏障穿透肽、细胞穿透肽等样本量仅有几百条的贫矿数据集上,传统深度学习和机器学习模型由于缺乏足够的训练数据,极易陷入过拟合的泥潭。而ESM-2凭借在海量通用蛋白质序列中积累的先验知识,即便冷冻了全部预训练参数、只用最简单的多层感知机作为预测头,依然能输出极具鲁棒性的预测结果。这说明,大模型确实已经学到了氨基酸排列背后的某种通用物理与演化规律。
其次,传统的理化描述符依然是性价比极高的防身利器。在传统机器学习的对比中,使用疏水性、电荷分布等物理化学描述符的模型,性能一致显著优于仅依赖氨基酸频率或One-hot编码的模型。这在直觉上非常合理——多肽的生物活性本质上是由其三维空间中的电荷与疏水表面决定的,这些人工构建的物理先验,为小模型提供了关键的底层约束。
然而,整篇工作最震撼、最具颠覆性的发现,在于对多肽同源聚类瓶颈的揭示。
在传统的蛋白质机器学习中,研究人员习惯于使用MMseqs2等经典比对工具对序列进行聚类,从而将同源序列隔离在训练集之外,防止数据泄漏。但在多肽这个短链世界里,团队发现这一套行之有效的规矩彻底失效了。无论如何优化聚类的相似度阈值和覆盖度参数,多肽序列聚类后的单序列簇比例始终高得离谱——绝大多数数据集的单序列占比都在40%以上,平均每个聚类的大小甚至不足3条序列。
这意味着什么?因为多肽序列太短,传统的比对算法根本无法将它们有效地组织成有意义的同源组,整个数据空间被切得极度碎片化。从下图的系统分析中可以清晰地看到这种过度碎片化的伪均匀分布特征,这直接导致了基于相似性划分的评估结果与随机划分几乎没有任何本质区别。
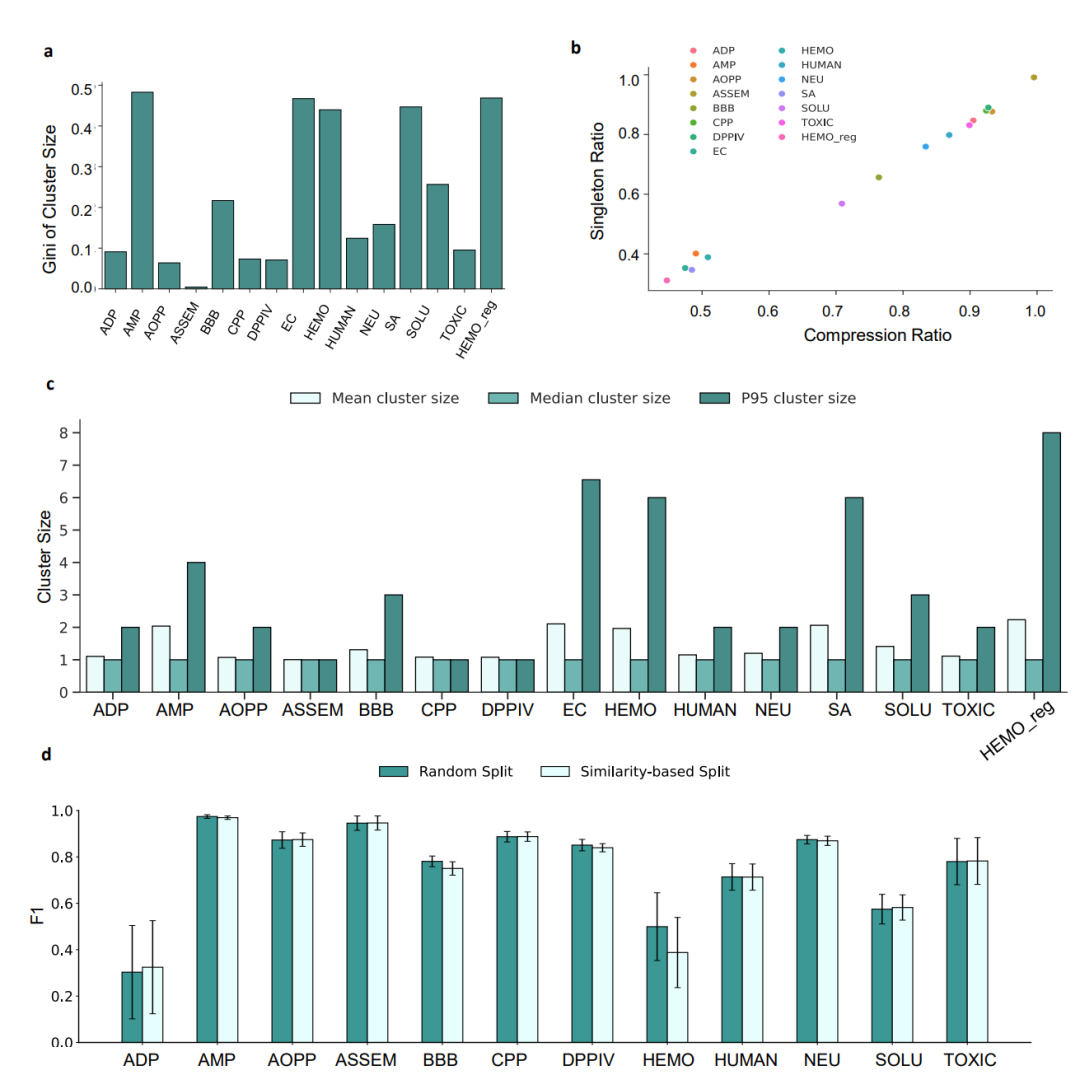
多肽聚类瓶颈分析:极高的单序列占比导致相似性划分与随机划分性能趋同
多肽聚类瓶颈分析:极高的单序列占比导致相似性划分与随机划分性能趋同
这一判断无疑给整个AIDD领域泼了一盆冷水。它无情地指出,过去大家自以为通过同源聚类划分测试集就能严谨评估多肽模型的泛化能力,实际上只是在一套被极度碎片化、本质上仍接近随机分布的数据集上进行了一场自我安慰。多肽的泛化评估,急需一种全新的同源性定义。
跨越一维序列天花板:下一代多肽AI制药的破局点在哪
研究团队的这项工作,不仅用详实的数据为多肽性质预测树立了行业标杆,更在行业展望中给出了极具穿透力的判断。
基于一维序列的多肽性质预测,或许已经逼近了其物理极限。多肽不同于拥有稳定折叠骨架的大型蛋白质,它们在溶液中具有极高的柔性,往往以构象系综的形式存在。当多肽与靶标蛋白结合时,通常会发生显著的诱导折叠。这意味着,仅仅依靠氨基酸排列的字符串信息,模型永远无法真正捕捉到多肽在动态结合过程中的药效团空间分布。
因此,下一代多肽预测工具的真正破局点,必然是跨越一维序列的阻隔,向三维结构与物理动力学靠拢。随着AlphaFold3、Boltz-2等能够精准预测多肽-蛋白质复合物三维结构与配体结合特征的通用模型相继开源,如何将动态构象采样、静电表面势能以及溶剂可及性等物理硬约束融入到性质预测流程中,将是决定AI能否真正闭环设计出高活性、低毒性、高稳定性多肽药物的关键。
团队在讨论中还明确提出了一个更深层的判断:多肽领域的同源性概念本身需要被重新定义。传统的全局序列比对在蛋白质世界里之所以有效,是因为蛋白质拥有足够长的序列来承载进化保守的结构域信息。但对于只有几个到几十个残基的多肽而言,真正决定其功能的可能只是某几个关键位置的药效团排布,或者某种局部的二级结构倾向性。未来的多肽泛化评估,或许需要从全局序列比对转向基于motif、药效团或局部结构相似性的全新度量体系。
为了让这些硬核发现真正沉淀为行业的公共财富,高琳与李鹏勇团队不仅开源了全部标准化数据集与评测代码,还开发了一个功能完备的在线服务平台 PPB Web Server⬇。在这个平台上,科研人员可以自由下载标准化数据集,交互式地可视化对比不同模型的表现,并直接调用其严谨的评估协议。
这项工作的边界也很清晰——它目前只触及了一维序列层面的预测,没有涉及非天然氨基酸修饰、环肽拓扑以及多肽-靶标复合物的三维相互作用。但正是这种诚实的边界意识,让整个基准的结论更加可信,也为后续的结构增强型基准留出了清晰的迭代空间。不搞噱头、脚踏实地为行业修路搭桥,这种开源精神,才是推动中国AIDD生态在多肽药物大潮中真正实现突破的核心动力。
参考文献
A Systematic Benchmark for Peptide Property Prediction
Dong Xiaoying, Yang Kaijun, Wu Tianxiang, Li Pengyong, Gao Lin
bioRxiv 2026.02.09.704773; doi: https://doi.org/10.64898/2026.02.09.704773
https://ppb.molmatrix.com/
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-31,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号