J. Med. Chem. | 「晶体结构优先」策略结合亿级化学空间对接:片段药物发现的范式革新

J. Med. Chem. | 「晶体结构优先」策略结合亿级化学空间对接:片段药物发现的范式革新

DrugIntel
发布于 2026-06-01 17:15:24
发布于 2026-06-01 17:15:24

文献来源: Müller J, Klein R, Tarkhanova O, et al. "Magnet for the Needle in Haystack: 'Crystal Structure First' Fragment Hits Unlock Active Chemical Matter Using Targeted Exploration of Vast Chemical Spaces." J. Med. Chem. 2022, 65, 15663–15678. DOI: 10.1021/acs.jmedchem.2c00813 合作机构: CrystalsFirst GmbH(德国马尔堡)· BioSolveIT GmbH · Enamine Ltd.(乌克兰基辅)· Chemspace LLC · 菲利普斯-马尔堡大学
一、研究背景与问题提出
1.1 传统苗头发现策略的局限
早期药物发现的核心任务是从化学空间中找到能与靶蛋白结合的"苗头化合物"(hit compounds)。当前主流策略包括:
- • 高通量筛选(HTS):将数十万至数百万化合物逐一测试活性,典型命中率仅约 1%;命中物分子量通常在 300–500 Da,结构复杂,后续优化空间有限,且对化学空间的覆盖仍然极为有限。
- • 虚拟筛选(Virtual Screening):通过分子对接预先过滤,降低实验成本;但传统对接方案在处理十亿量级以上化合物库时,计算代价依然高昂。
1.2 片段药物发现(FBDD)的崛起与瓶颈
FBDD 以分子量通常低于 300 Da、重原子数少于 20 个的"片段"分子为起点,其核心优势在于:
- • 小分子与靶蛋白的结合更加高效(配体效率 LE 更高),更易发现关键相互作用热点;
- • 更系统地覆盖化学空间,增加找到全新骨架的概率;
- • 已成功推动 sotorasib(KRAS G12C 抑制剂)、asciminib(BCR-ABL 抑制剂)等药物获批上市,并在"不可成药"靶点(如 RAS、KEAP1)上展现出独特优势。
然而,FBDD 传统路径中的生物物理预筛选级联存在严重缺陷。多项研究(Schiebel et al., 2016;Chang et al., 2021)表明,不同生物物理方法(SPR、ITC、NMR、TSA 等)在平行筛选中所识别片段的重叠率极低,大量真实结合片段在进入 X 射线结晶验证之前便已被过滤丢弃。这意味着以活性/亲和力为导向的预筛选策略,本身就存在系统性漏报风险。
1.3 本文的核心命题
本文针对上述问题,提出并验证了一套以 X 射线晶体结构为唯一起点、彻底绕过生物物理亲和力预筛选的全新工作流——"Crystal Structure First"(晶体结构优先),并将其与 Enamine 26 亿分子量级的 REAL Space 化学空间对接相结合,实现从结构确认的片段到纳摩尔级活性化合物的高效跨越。
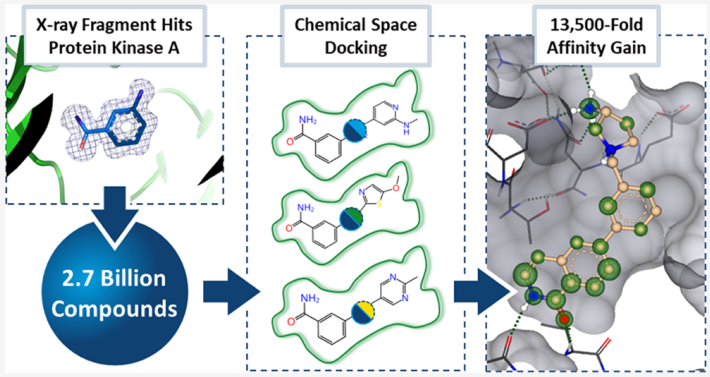
二、研究体系与靶标选择
2.1 靶蛋白:蛋白激酶 A(PKA)
PKA(cAMP 依赖性蛋白激酶)是研究最为深入的丝/苏氨酸激酶之一,其 ATP 结合位点(催化区)是本研究的作用靶点。研究团队选择 PKA 的主要原因在于:
- • 已有由 Oebbeke et al. 和 Siefker 完成的大规模晶体学片段筛选数据库(19 个可靠片段复合物结构可供利用);
- • 铰链区(hinge region)结合模式明确,利于系统评估片段骨架多样性;
- • 功能性磷酸化测定体系成熟,可提供定量 Ki 值。
2.2 起始片段的选择原则
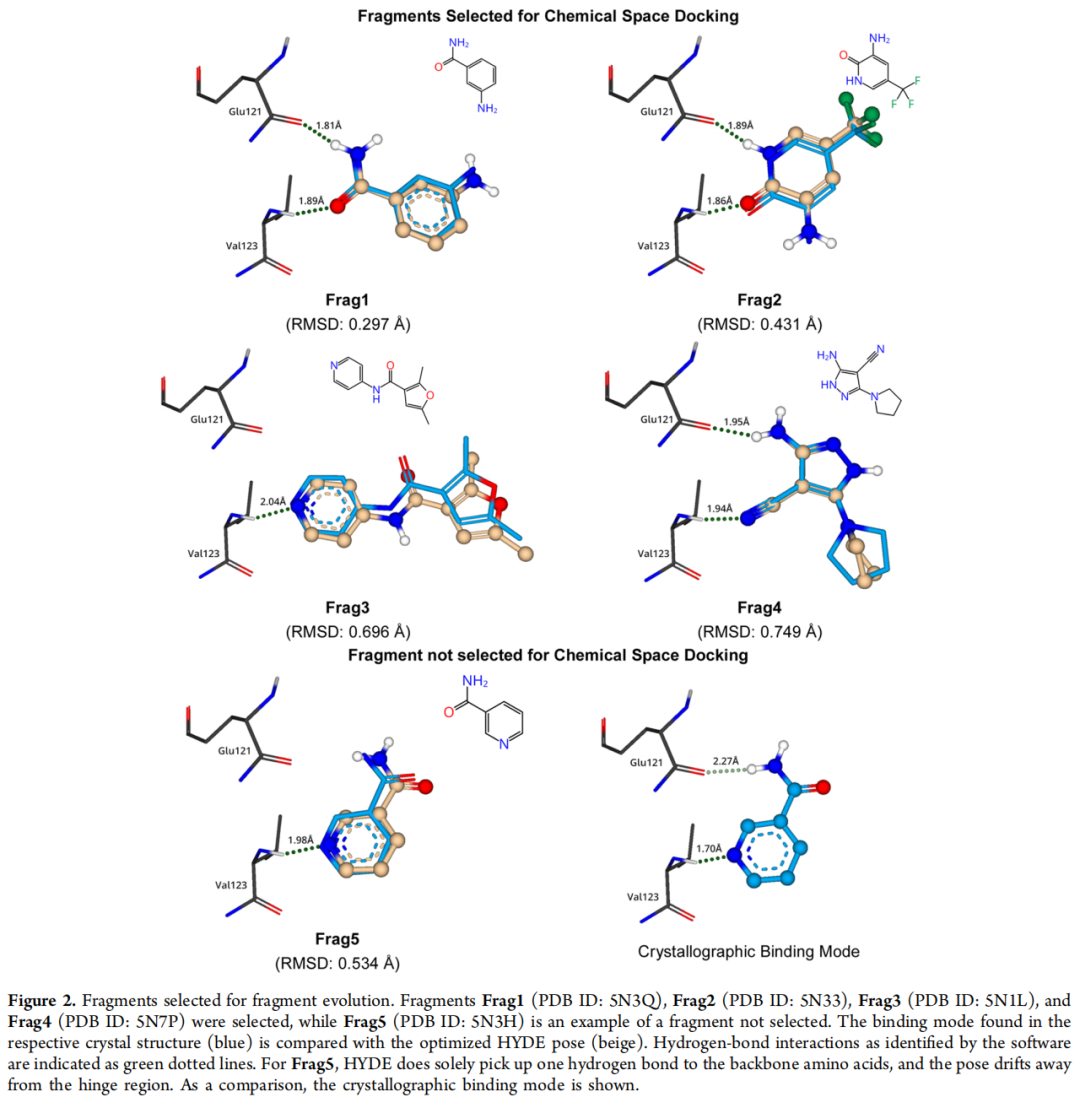
从 19 个高可信度晶体结构(依据配体密度质量、占有率、RMSD 等标准筛选)中,仅凭结合模式,不考虑任何亲和力数据,选出 4 个化学多样性良好的铰链区结合片段:
片段 | PDB ID | 关键药效基团 | HYDE 优化后 RMSD |
|---|---|---|---|
Frag1 | 5N3Q | 伯酰胺(primary amide) | 0.297 Å |
Frag2 | 5N33 | 内酰胺(lactam) | 0.431 Å |
Frag3 | 5N1L | 吡啶(pyridine) | 0.696 Å |
Frag4 | 5N7P | 伯胺/腈基(primary amine/nitrile) | 0.749 Å |
评估工具: HYDE 评分函数(SeeSAR v10.1)——首先对片段姿态进行氢键几何、分子内构象应变和空间碰撞的预优化,再估算结合自由能。RMSD 偏差小的片段表明晶体学结合模式与力场优化结果高度一致,可信度更高。 对照被排除的 Frag5(PDB: 5N3H):HYDE 优化后姿态偏离铰链区,与晶体结合模式存在显著差异,予以排除。
三、化学空间对接工作流(详细拆解)
整个计算流程分为三个串联步骤,形成从晶体片段到亿级候选分子再到精选合成列表的完整漏斗。
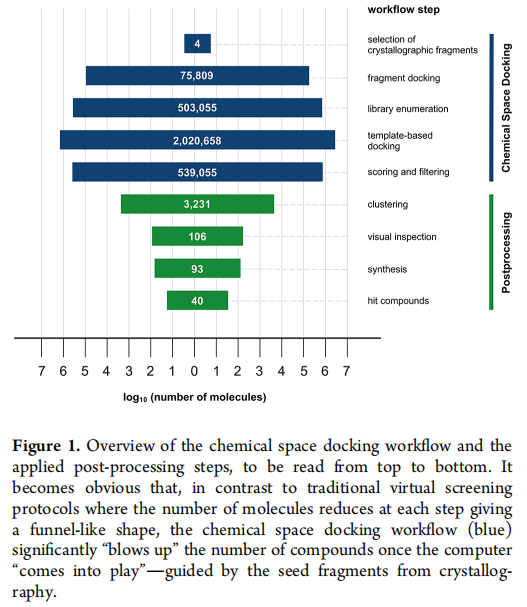
3.1 步骤一:REAL Space 合成砌块的模板对接
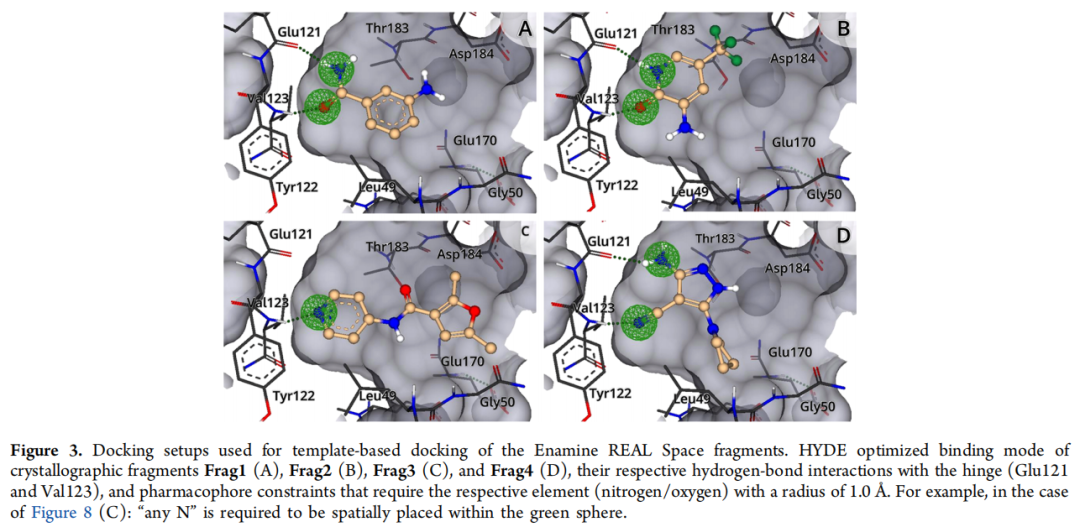
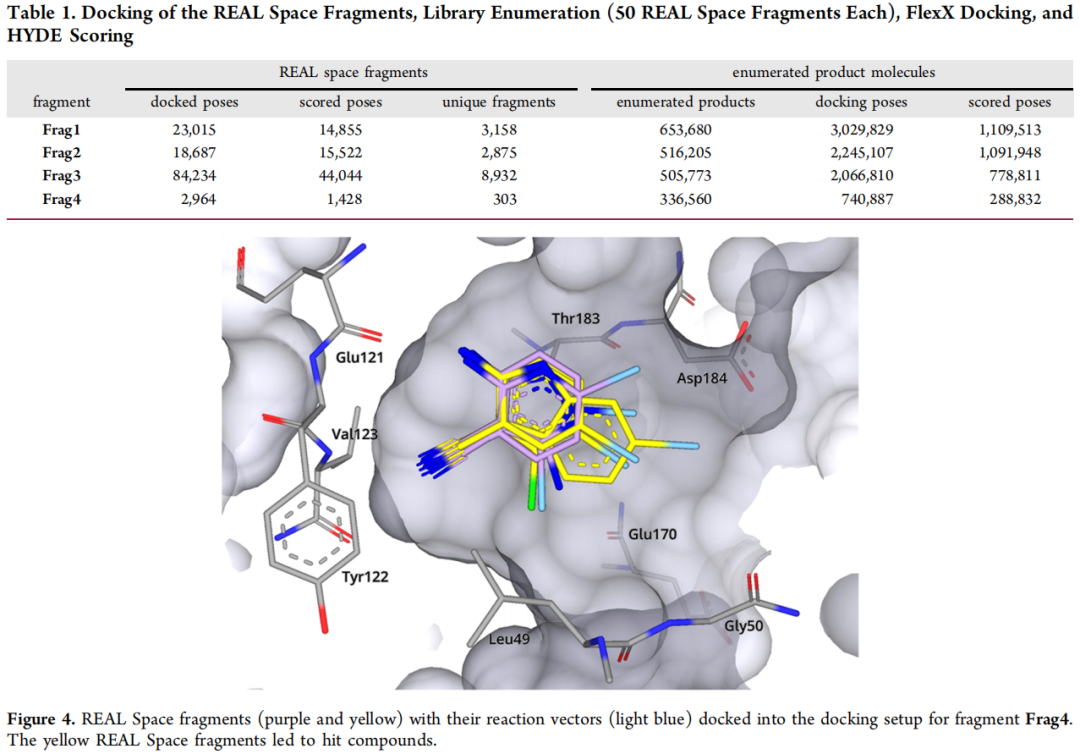
目标: 将 Enamine REAL Space 中所有可用的反应砌块("synthons",即虚拟合成子,共 208,293 个)对接至以晶体片段坐标为模板的结合位点,筛选空间位置匹配的砌块。
核心算法:FlexX(SeeSAR 集成版本)基于最大公共子结构(MCS)的模板对接
- • 每个砌块生成 10 个姿态,要求与晶体片段存在至少 5 个原子的"模糊"MCS 匹配("fuzzy"——例如允许脂肪族氮匹配芳香族氮),引入约 0.5–1.5 Å 的构型灵活性,增加命中多样性。
- • 设置原子级药效团约束(半径 1.0 Å):要求特定氮或氧原子精确落入铰链区氢键供/受体位置(Glu121 和 Val123 骨架)。
- • HYDE 后评分保留前 50,000 个姿态,每个片段组进一步筛选 50 个 REAL Space 砌块用于后续枚举,选择标准为 MCS 大小、反应向量方向、低扭转能及配体效率(LE ≥ 0.3)。
产出: 每个晶体片段对应约 300–8,900 个唯一匹配砌块(Frag3 因吡啶环在 REAL Space 中高度表征而数量最多)。
3.2 步骤二:基于反应知识的子库枚举
目标: 利用 Enamine 专有的反应知识库,将步骤一筛选出的 50 个 REAL Space 砌块,按照其各自携带的"反应向量"(reaction vector)枚举成完整的二组分反应产物。
- • 使用 CoLibri(BioSolveIT)执行"反应感知"枚举,确保所有枚举产物在化学上均可合成;
- • 每个砌块平均对应约 13,000 个虚拟产物;
- • 每个晶体片段最终枚举出约 503,055 个化合物的子库,四个片段合计约 201 万个候选分子。
本研究使用的化学空间版本(REAL Space 2020-07,两组分反应)基于 173 种反应和 110,269 个砌块,理论上覆盖 26.57 亿个虚拟产物。
3.3 步骤三:枚举子库的模板对接与评分
目标: 对步骤二产出的约 200 万化合物进行第二轮对接,此次以步骤一中各砌块的对接姿态为模板(而非晶体片段),保留整个分子的扩展方向。
- • 每个分子生成 5 个姿态,HYDE 评分过滤;
- • 蛋白表面空间碰撞检测和构象应变过滤共淘汰约 50–60% 的姿态;
- • 每个片段组最终保留约 539,055 个打分姿态用于后处理。
- • 全程计算耗时平均仅约 6 小时(标准化至 1,000 CPU 核),相比传统全库对接节省约 10 倍计算资源。
3.4 后处理:新颖性验证与聚类精选
- 1. 新颖性验证: 从 ChEMBL 和 PDB 提取 131 个已知 PKA 活性化合物(IC₅₀ < 1 µM),计算与候选化合物的 Tanimoto 相似性(Morgan 指纹)。各簇最高相似性为 0.452–0.547,确认候选化合物与已知抑制剂的化学骨架差异显著。
- 2. Tanimoto 聚类: 使用 RDKit(KNIME 平台)进行 2D Morgan 指纹聚类,过滤非类药性质(铅样分子过滤),最终得到 3,231 个化合物(c1: 1,342;c2: 816;c3: 607;c4: 466)。
- 3. 人工目视检查: 评估氢键网络(重点关注 Lys72、Glu127 侧链及 Phe54/Leu74 疏水口袋)、疏水互补性、环构象合理性及扭转角分布(torsion histograms),最终选定 106 个化合物委托合成。
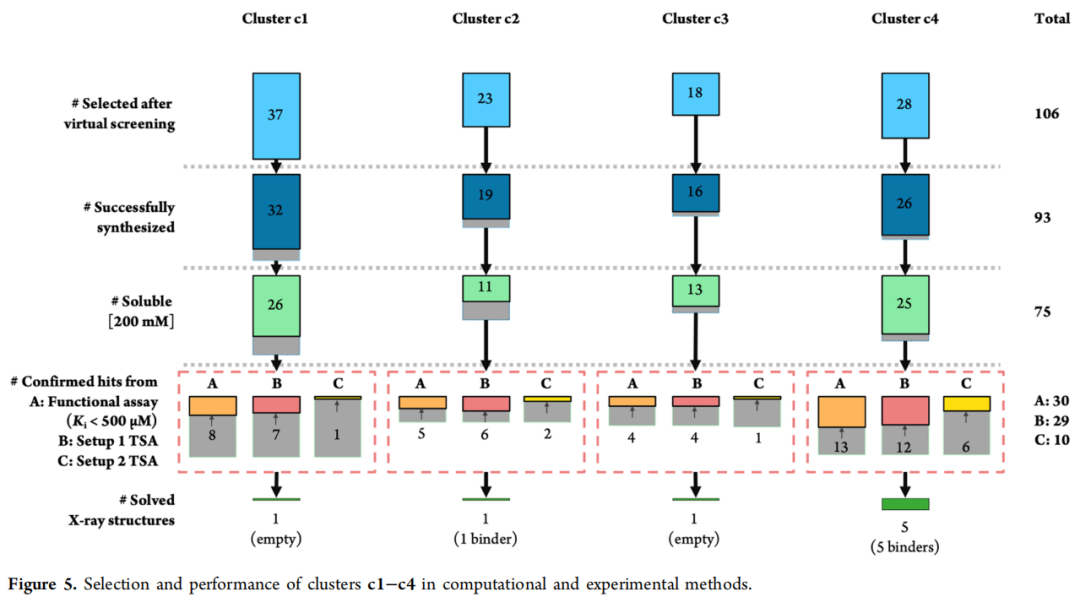
四、化合物合成与活性评估
4.1 并行化学合成
106 个候选物经 Enamine 按需合成(Make-On-Demand),成功合成 93 个化合物(合成成功率 88%),所有化合物纯度 ≥ 95%(LC/MS 或 ¹H NMR 确认)。
涉及的主要合成转化类型(共 21 种程序):
转化类型 | 代码 | 典型应用簇 |
|---|---|---|
Suzuki 偶联 | T1 | c2, c4 |
酰胺偶联 | T2 | c1, c2 |
脲形成 | T3 | c3 |
烷基化/还原胺化 | T4 | c1, c3 |
芳基化 | T5 | c4 |
磺酰胺形成 | T6 | c1, c4 |
4.2 功能性激酶磷酸化测定
采用商业化 Z'-LYTE 激酶测定试剂盒(Invitrogen/Thermo Fisher,Ser-Thr 1 肽),以荧光比率法定量磷酸化肽的生成,通过 Cheng–Prusoff 方程(ATP KM = 3.8 µM)将 IC₅₀ 换算为 Ki 值,三重复测定。
关键结果:
- • 75 个化合物可溶于 200 mM DMSO 并完成测定;
- • 40 个化合物(40%)在功能测定中显示抑制活性;
- • 最优化合物 EN093(c2 簇,外环 S-构型)Ki = 744 nM,对应的初始片段 Frag2 活性仅为约 10 mM,亲和力提升 13,500 倍;
- • 多个化合物展现出清晰的浓度-效应关系和良好的 LE(0.20–0.35)。
4.3 热位移实验(TSA)交叉验证
为与工业界常规筛选流程进行直接比较,在两种条件下对所有化合物进行 TSA:
- • Setup 1(高浓度):配体 2.5 mM,约 340 倍蛋白摩尔量过量;29 个化合物显示显著 Tm 变化(22 正移 + 7 负移),其中 18/22 阳性移动化合物在功能测定中有活性。
- • Setup 2(低浓度):配体 70 µM,10 倍蛋白摩尔量过量(模拟工业标准筛选条件);仅 10 个阳性移动化合物均在功能测定中有活性。
关键发现(对 FBDD 传统范式的直接挑战): 将四个初始片段(Frag1–4)在相同条件下测定:
- • Setup 2 条件下,所有片段的 ΔTm 均 < 1.0°C,均未达到显著性阈值;
- • Setup 1 高浓度条件下,仅 Frag4 显示 1.6°C 位移(仍属弱信号)。
这意味着,若按照工业界标准 TSA 流程进行预筛选,这四个片段将全部被排除,后续所有活性化合物的发现将无从实现。
4.4 碳酸酐酶(CA)选择性对照实验
对 88 个化合物进行 CA(牛红细胞来源)TSA 筛选,验证对 PKA 的选择性。仅 EN020(含芳基磺酰胺结构)显示弱 CA 活性,而 EN020 对 PKA 无活性——确认其余活性化合物对 PKA 的结合具有特异性,排除非特异性聚集等假阳性干扰。
五、X 射线共晶结构解析与结合模式验证
5.1 共晶实验设计与成功率
选取各簇最活跃化合物共 13 个进行共结晶(sitting-drop 蒸汽扩散法,4°C,18–23% 甲醇沉淀剂),数据在 DESY(汉堡,P11 束线)和 BESSY II(柏林,14.1 束线)收集,100 K,波长 1.033 Å。
- • 13 个化合物中 9 个获得可用晶体(70% 成晶率);
- • 8 个数据集质量足够精修;
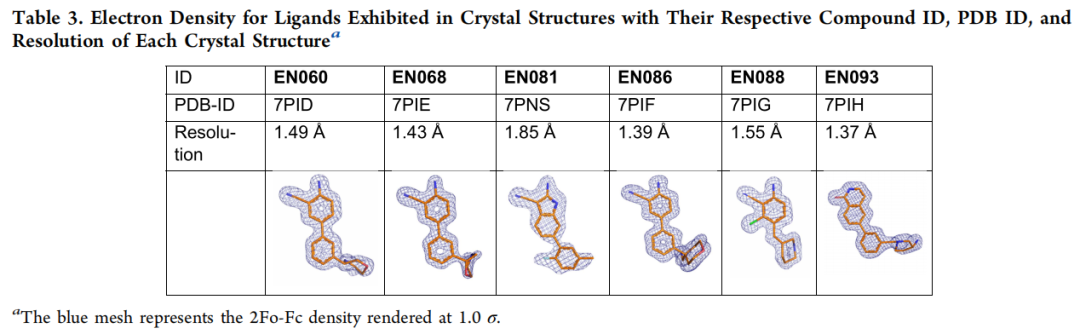
- • 6 个化合物获得明确的配体结合共晶结构(分辨率 1.37–1.85 Å,PDB: 7PID/7PIE/7PNS/7PIF/7PIG/7PIH)。
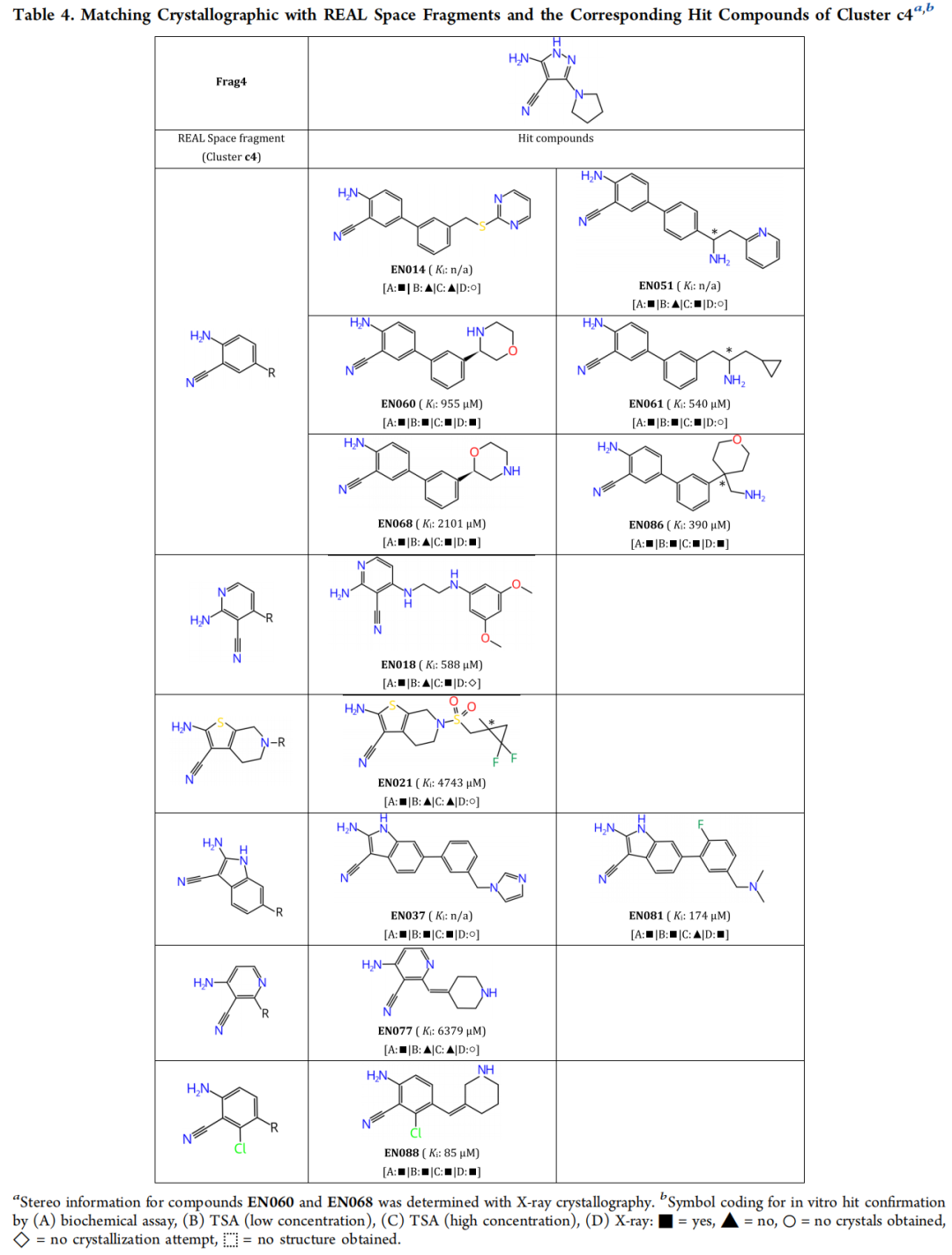
5.2 c4 簇:吡唑-4-腈系列
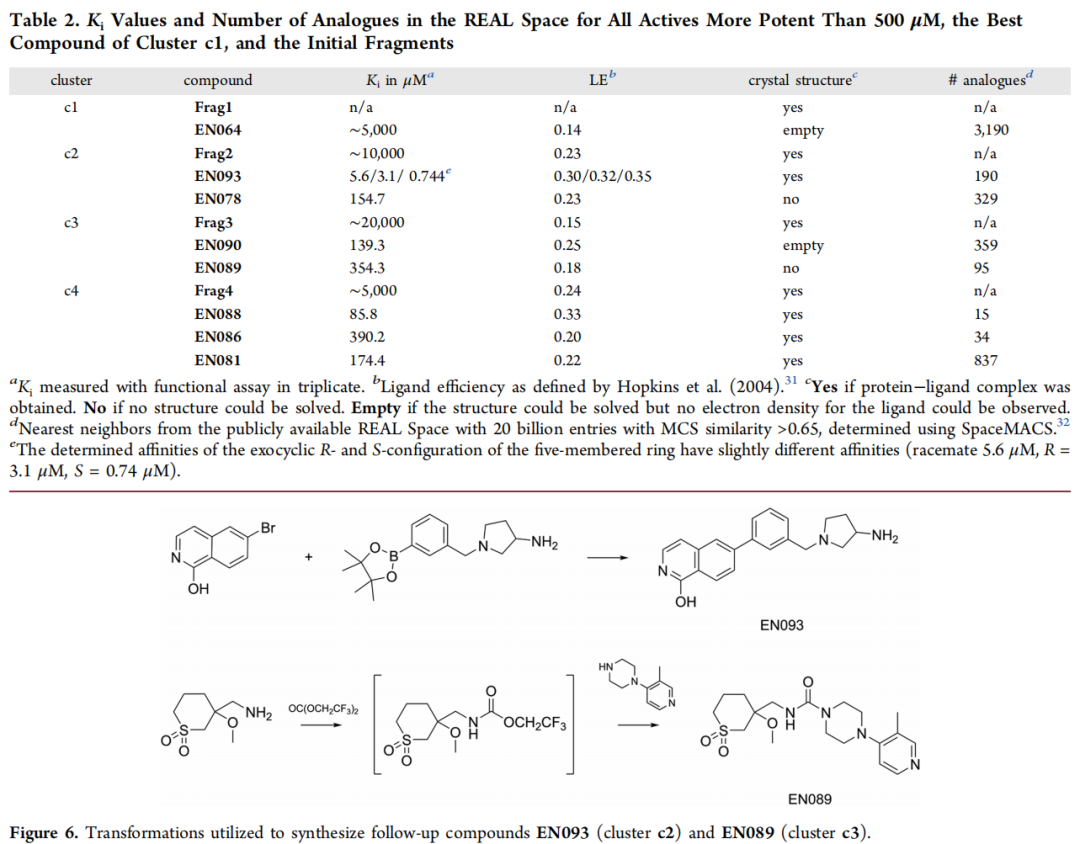
命中化合物与关键结构特征(依据晶体结构):
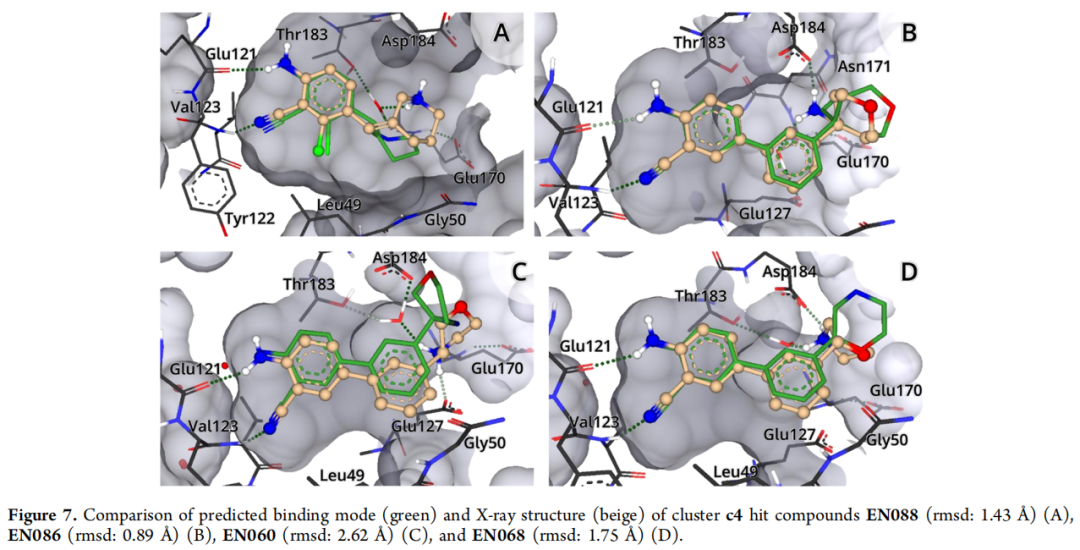
化合物 | Ki | PDB | RMSD(对接 vs 晶体) | 关键相互作用 |
|---|---|---|---|---|
EN088 | 85 µM | 7PIG | 1.43 Å | 氯原子填充 Leu49/Leu173/Phe327 疏水口袋;哌啶仲胺水媒介 H 键至 Glu170/Thr183 |
EN086 | 390 µM | 7PIF | 0.89 Å | 伯氨基多重 H 键至 Asn171/Asp184;水媒介 H 键至 Glu127/Glu170;四氢吡喃朝向溶剂 |
EN060 | 955 µM | 7PID | 2.62 Å | 吗啉氮 H 键至蛋白;水媒介 H 键至 Thr183/Asp184 |
EN068 | 2101 µM | 7PIE | 1.75 Å | 吗啉氮直接 H 键至 Asp184;六元脂肪环构象较灵活(占有率 87%) |
EN081 | 174 µM | 7PNS | 1.52 Å | 吲哚部分相对初始片段 Frag4 有扭转,铰链 H 键受影响;二甲氨基电子密度无法解析 |
模糊算法的重要作用: c4 簇的 5 个命中化合物均包含六元脂肪环作为铰链结合基元,而 Frag4 本身携带的是五元吡唑环。这种"五元→六元"环扩展的匹配,正是由 FlexX 的模糊 MCS 算法实现的,体现了算法在结构多样性探索上的优势。
5.3 c2 簇:异喹啉系列(最优化合物 EN093)
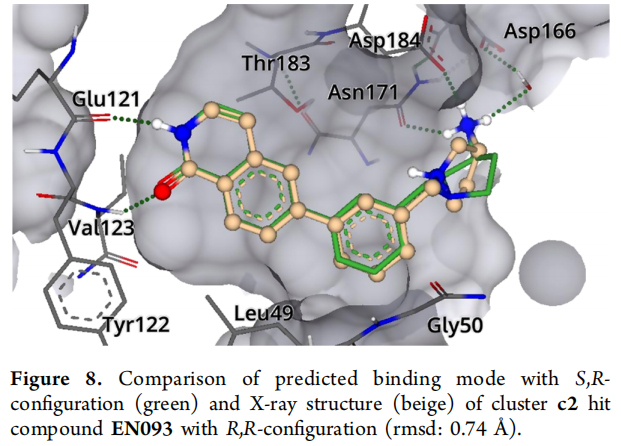
EN093 是本研究活性最强的化合物,其共晶结构(PDB: 7PIH,分辨率 1.37 Å)揭示了以下精细结合模式:
- • **伯氨基(很可能质子化)**与 Asn171 和 Asp184 侧链形成直接氢键;
- • 水媒介氢键至 Asp166 侧链;
- • FlexX 对接算法预测的伯氨基位置与晶体结构 RMSD 仅 0.74 Å,高精度验证计算预测;
- • 分子包含两个立体中心:内环氮(可在溶液中快速差向异构)和外环碳原子(化学稳定)。
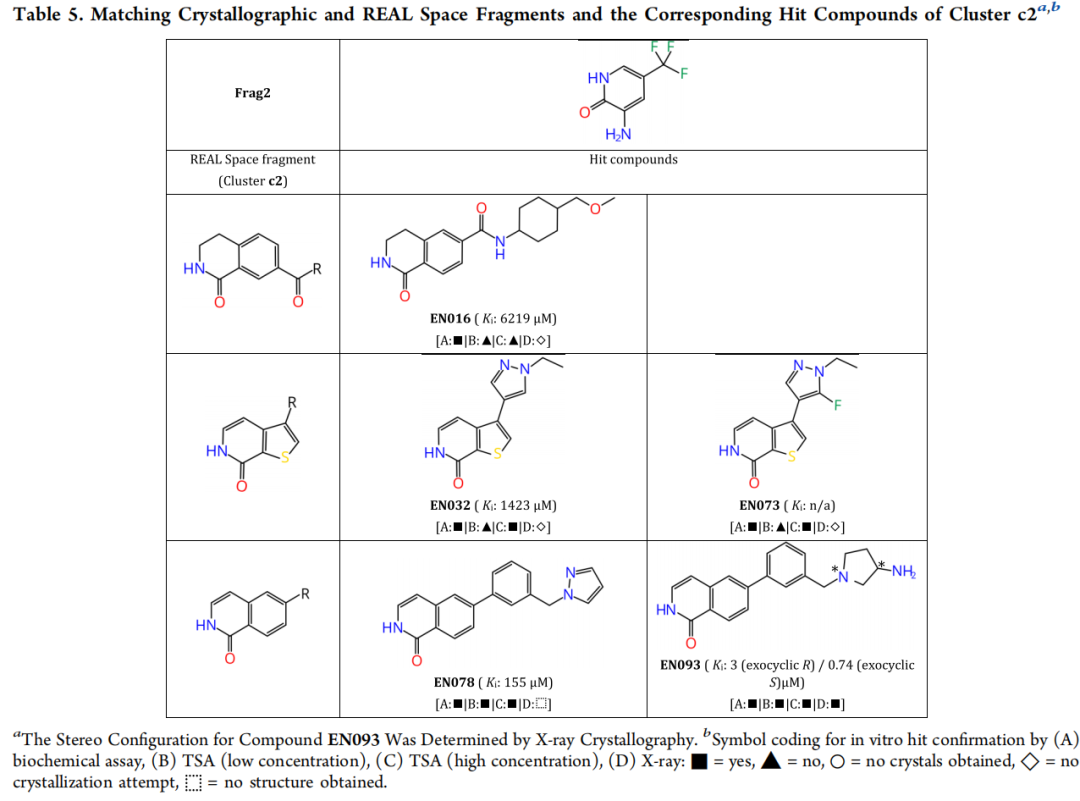
立体化学的精细分析: 对接模型预测 S,R-构型;晶体结构(外消旋体结晶)显示 R,R-构型为主要物种。分别合成 R- 和 S-构型对映体后的功能测定表明:S-构型(744 nM)略优于 R-构型(3.1 µM),考虑到测定精度,两者差异约为 4 倍。这为后续消旋稳定化修饰和立体选择性合成路线的 SAR 研究提供了重要线索。
六、整体成效与方法学反思
6.1 核心指标汇总
指标 | 数值 |
|---|---|
化学空间规模 | 26.57 亿虚拟产物 |
初始晶体片段数 | 4 |
对接候选物(筛后) | 3,231 |
委托合成 | 106 |
合成成功 | 93(88%) |
功能测定活性(Ki < 500 µM) | 40(40%) |
共晶结构解析 | 6 |
最大亲和力提升 | 13,500 倍(片段 → EN093) |
总周期 | 9 周 |
计算资源节省 | 约 10 倍(vs 传统全库对接) |
6.2 与 HTS 的本质差异
传统 HTS 通常以 1% 命中率获取 µM 级苗头,化合物分子量 300–500 Da,后续需要大量 SAR 优化,且结构信息稀缺。本研究以 40% 命中率、在 9 周内获得多个 nMµM 级化合物,且全部伴随清晰的 SAR 脉络和至少部分共晶结构——是对 HTS 模式的有力补充乃至替代。
6.3 TSA 范式局限的实证
本研究最具挑战性意义的发现之一:所有初始片段在工业界标准 TSA(低浓度 setup 2)条件下均未被检出,这与文献中记录的多种生物物理方法之间命中重叠率低的现象一致。作者将此视为直接证据,支持"预筛选级联本身会系统性遗漏片段命中物"的论断,并为"晶体结构优先"路径的合理性提供了最有力的注脚。
6.4 方法的优势与局限
优势:
- • 以经过晶体结构验证的结合模式直接驱动计算设计,大幅减少假阳性;
- • 基于 REAL Space 可预测反应路径的化学空间保证高合成成功率;
- • 计算效率高,对大型化学空间的覆盖兼顾深度与广度;
- • 一次迭代即可从毫摩尔级片段跃升至亚微摩尔级苗头,时间线极短;
- • 模糊 MCS 算法允许一定程度的结构跃迁(scaffold hopping),增加化学多样性。
局限与前提:
- • 依赖高质量蛋白结晶条件:靶蛋白需能与片段共结晶并产生可分辨的电子密度,对蛋白表达、纯化和结晶方案有较高要求;
- • 片段数量有限:本研究仅使用 4 个片段,统计代表性较为有限,作者亦承认这一点;
- • 未在第一轮迭代考虑选择性:目前工作流聚焦于 PKA 结合活性,对激酶家族选择性的系统评估尚未纳入流程;
- • 化学空间版本的时效性:REAL Space 处于持续扩展中(2020-07 版本 26 亿 → 现已超 200 亿),工作流本身可直接受益于库的扩大。
七、展望与实际应用价值
7.1 SAR-by-Space 策略的延伸
作者指出,本研究获得的化合物和结构数据,可直接作为第二轮"化学空间对接"的输入,通过 Tanimoto 相似性阈值的调整,系统性地探索各活性骨架周围的 SAR。EN093(c2 簇)的伯氨基-Asn171/Asp184 氢键锚定模式,以及 EN088(c4 簇)的氯取代疏水填充模式,均为下一轮优化提供了清晰的结构基础。
7.2 对"不可成药"靶点的潜在意义
本文引用了 KEAP1(Astex)和 pan-RAS(Boehringer Ingelheim)的案例,说明在蛋白质-蛋白质相互作用(PPI)和浅表结合口袋等传统 HTS 难以奏效的场景中,"晶体结构优先"策略的价值尤为突出。将本文工作流与 Pan-RAS 等靶点的大规模晶体学片段筛选(如 XChem 平台)相结合,是一个值得期待的研究方向。
7.3 "三角"工作流的协同效应
作者以三个相互支撑的能力概括了本方法的成功要素:
(a) 高质量晶体学片段命中物的高效获取
↕
(b) 亿级化学空间的定向子枚举与计算筛选
↕
(c) 基于可预测反应知识的按需合成这三者的协同,使得从结构到活性分子的"最后一公里"变得可预期、可复现、可扩展,为制药行业早期发现阶段提供了一套经过实验验证的系统性解决方案。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号