开源平台赋能多靶点结构-活性关系研究

开源平台赋能多靶点结构-活性关系研究

DrugIntel
发布于 2026-05-29 12:59:35
发布于 2026-05-29 12:59:35

原文信息 Espinoza-Castañeda J.I., de la Fuente-Nunez C., Medina-Franco J.L. Open-source chemoinformatics platforms for multi-target structure-activity relationships Cell Reports Physical Science, 7, 103276, May 20, 2026 DOI: 10.1016/j.xcrp.2026.103276 代码仓库:github.com/IsrC11/SMARTs-Toolbox
目录
- 1. 背景与研究动机
- 2. 核心概念辨析:SAR → SMARTs → 多药理学
- 3. 工具选择标准与分类框架
- 4. 四大分析类别详解
- • 4.1 数据可视化工具
- • 4.2 相似度预测工具
- • 4.3 定量分析工具(QSAR/multi-QSAR)
- • 4.4 机器学习与深度学习工具
- 5. 商业工具概览与开源工具对比
- 6. 按编程能力分层的工具选择矩阵
- 7. 实战案例:短肽多靶点设计中的 SMARTs 集成工作流
- 8. 现存局限与关键挑战
- 9. 未来展望
1. 背景与研究动机
1.1 单靶点范式的局限性
结构-活性关系(Structure-Activity Relationships, SAR)是药物化学与先导化合物优化的核心方法论,其本质是通过统计或模型方法,揭示分子结构特征(描述符、骨架、片段)与生物/理化活性之间的定量关联。然而,传统 SAR 的根本假设是"一药一靶"——即一个分子只针对一个治疗靶标进行设计与优化。
这一还原论范式在面对复杂多因素疾病时正面临越来越严峻的挑战:
- • 癌症:涉及 PI3K/AKT/mTOR、RAS/MAPK、Wnt/β-catenin 等多条信号通路的交叉激活,单靶点抑制极易诱导旁路补偿机制;
- • 阿尔茨海默病:Aβ 聚集、tau 蛋白过磷酸化、神经炎症及胆碱能功能障碍协同作用,任何单一干预均难以逆转整体病理进程;
- • 多重耐药感染:病原体可通过靶标突变、外排泵上调或生物膜形成等机制绕过单一作用机制的药物。
与此同时,化学、生化及生物物理数据的爆炸式增长(大规模表型筛选、高通量测序、蛋白质组学)使整合多来源信息变得既必要又可行。
1.2 多药理学与 SMARTs 的兴起
多药理学(Polypharmacology)是指单一分子对多个蛋白靶标产生有意义相互作用的能力,这既可能是设计意图,也可能源于意外的脱靶效应。区分"有益的多药理学"与"有害的滥靶性(promiscuity)"是当前领域的核心挑战之一。
在此背景下,结构-多活性关系(Structure-Multiple Activity Relationships, SMARTs) 作为 SAR 的系统性扩展应运而生。SMARTs 不再局限于单靶点活性数据,而是通过整合多靶点、多组学、多通路数据,同时优化化合物在若干治疗维度上的活性谱。
已获批的多靶点药物为这一策略提供了有力的临床证明:
药物 | 适应症 | 已知多靶点机制 |
|---|---|---|
索拉非尼(Sorafenib) | 肝癌/肾癌 | RAF激酶、VEGFR、PDGFR等多激酶抑制 |
多奈哌齐(Donepezil) | 阿尔茨海默病 | AChE抑制 + 5-HT受体调节 |
伊马替尼(Imatinib) | CML | BCR-ABL、c-Kit、PDGFR多靶点抑制 |
然而值得警惕的是,目前有意设计的多靶点批准药物仅占全部上市药物的约 15%,反映出从计算设计到临床转化之间依然存在巨大鸿沟。
2. 核心概念辨析
SAR vs. SMARTs
SAR(结构-活性关系)
├── 输入:一个化合物集合 × 一个生物活性端点
├── 输出:结构特征 → 单一活性的定量模型
└── 局限:忽略靶标间的相关性与化合物的多维活性谱
SMARTs(结构-多活性关系)
├── 输入:一个化合物集合 × 多个靶标/通路/性质端点
├── 输出:结构特征 → 多维活性谱的整合模型
└── 优势:支持多参数同步优化,识别跨靶标的共性结构片段SMARTs 与药物重定向
SMARTs 分析的一个重要应用场景是药物重定向(Drug Repurposing):通过识别不同治疗靶标之间共享的结构基序(motif),预测已批准化合物在新适应症中的潜力,显著压缩早期发现成本。
3. 工具选择标准与分类框架
3.1 文献检索策略
作者对 PubChem、GitHub 和 Google Scholar(2020–2025年)进行系统检索,关键词涵盖:
- •
chemoinformatics+open access - •
structure-activity relationships+application note - •
polypharmacology+polypharmacology tools - • 相似度引文追溯(以 SARANEA 为种子文献)
3.2 工具入选标准(优先级排序)
- 1. 多靶点适用性:有文献记录的或潜在的多靶点应用场景;
- 2. 时效性与可获取性:功能已于2026年验证,具备稳定访问入口;
- 3. 可集成性:能与标准化学信息学工作流直接对接;
- 4. 代表性与验证度:兼顾新兴工具与经社区广泛验证的成熟平台。
3.3 分类框架:功能维恩图
本综述将15个平台(10个开源 + 5个商业)划分为四大功能类别,并以维恩图呈现多功能交叠关系:
┌─────────────────────────────────────┐
│ 数据可视化(Data Viz) │
│ ChemGPS · MAYA · webDrugCS │
│ DataWarrior · SARvision │
│ ┌──────────────┐ │
│ │ Flare │ │
└──────────┤ (Viz+Quant) ├────────────┘
│ Optibrium │
┌───────────────┤ (Viz+Quant ├────────────────┐
│ 定量分析 │ +ML) │ 相似度预测 │
│ SARANEA └──────────────┘ 3DSTarPred │
│ DTINet ↑ OpenEye │
│ ┌─────────────┐ │
└────────────┤ Chemprop ├───────────────────┘
│ (Sim+ML) │
│ Polygon │
│ OpenChem │
│ DeepMol │
│ DeepAutoQ │
└─────────────┘
机器学习(ML)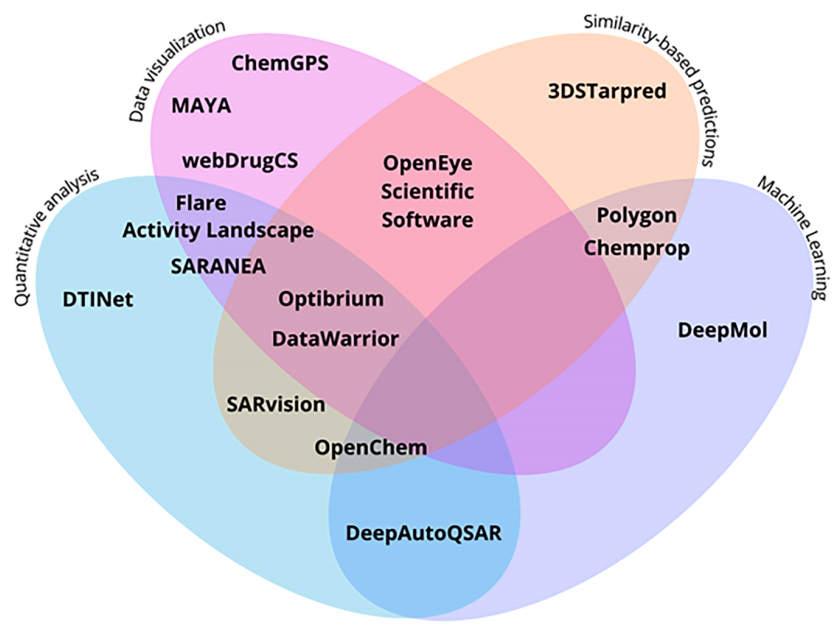
图1. 化学信息学工具。依据工具在药物发现中的多重作用,以维恩图形式进行组织与展示,包括数据可视化(粉色)、基于相似性的预测(橙色)、定量分析(蓝色)以及机器学习(紫色)。各工具被放置于其对应的功能类别中,重叠区域则表示其具备多功能特性
4. 四大分析类别详解
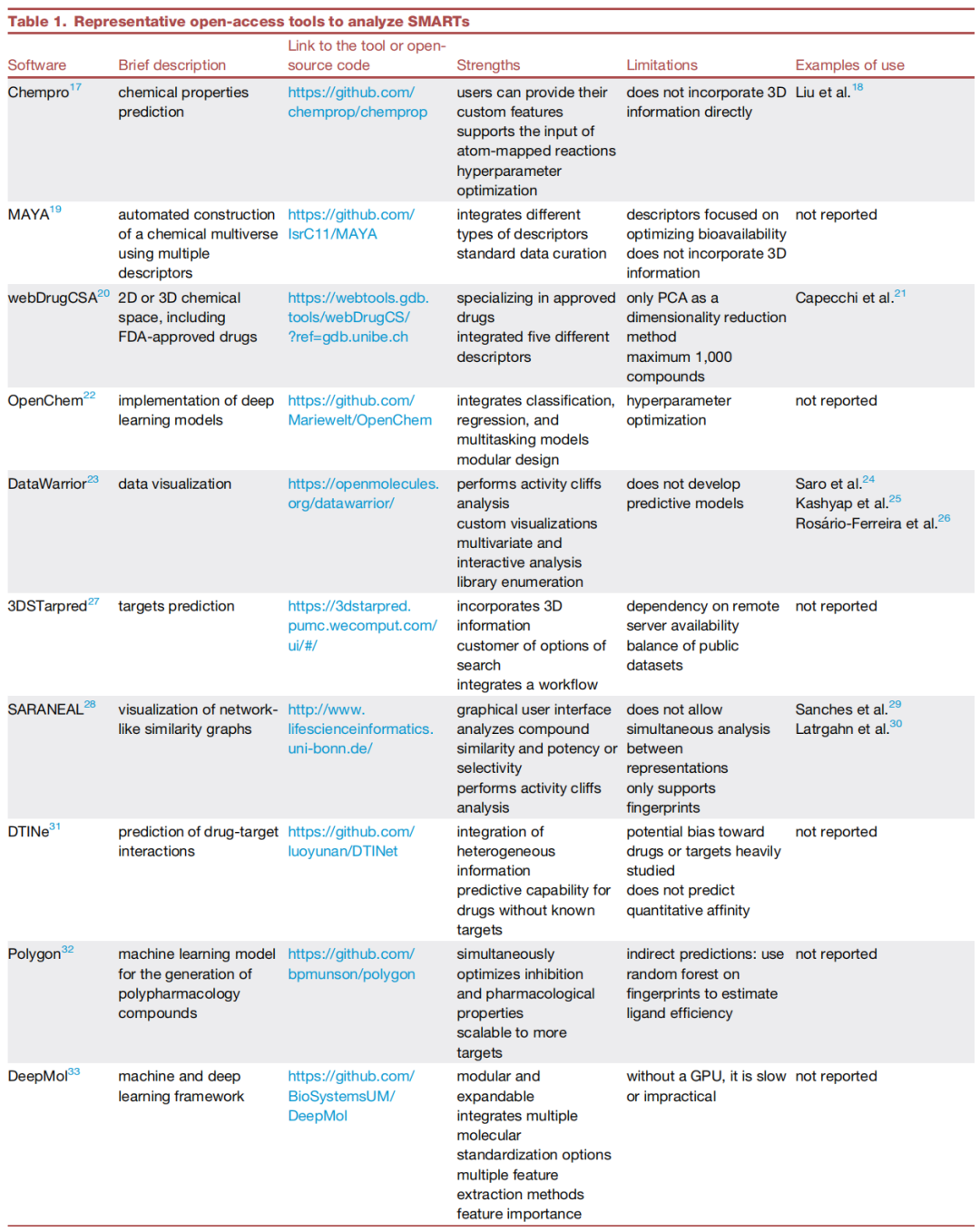
4.1 数据可视化工具
数据可视化在 SMARTs 分析中承担化学空间导航的核心角色,通过将高维描述符映射到可视化二维空间,揭示化合物在多活性维度上的聚类、离群与活性悬崖(Activity Cliff)模式。
MAYA(Multiple ActivitY Analyzer)
- • 开发语言:Python
- • 代码仓库:github.com/IsrC11/MAYA
- • 核心功能:
- • 自动化构建"化学多元宇宙"——对同一化合物集合同时生成基于不同描述符类型(理化性质、结构指纹)的多组互补化学空间表示;
- • 支持三种降维算法:PCA(线性,全局结构保留佳)、UMAP(非线性,局部结构+全局拓扑)、t-SNE(非线性,局部聚类保留佳);
- • 内置标准数据清洗(Data Curation)流程;
- • 整合多类型描述符(理化描述符、MAP4指纹等)。
- • 适用场景:需要深度探索 SMARTs 模式的技术用户,适合同时处理多活性标注数据集。
- • 主要局限:
- • 缺乏专属图形界面(GUI),非专业用户上手曲线较陡;
- • 描述符集中于优化生物利用度特征,暂不支持三维构象信息。
DataWarrior
- • 类型:开源桌面软件(Java)
- • 获取地址:openmolecules.org/datawarrior
- • 核心功能:
- • 交互式化学空间可视化(2D/3D)
- • 活性悬崖分析(基于相似度矩阵与活性差异阈值)
- • 分子描述符计算与多变量分析
- • 化合物库枚举与文库设计
- • 完全图形化操作,无需编程
- • 适用场景:无编程背景的研究人员,日常化学数据探索与分析。
- • 主要局限:不具备构建预测模型的能力,缺乏多靶点活性的定量建模支持。
webDrugCS
- • 获取地址:webtools.gdb.tools/webDrugCS
- • 特色:内置FDA批准药物化学空间数据库,集成五种不同分子描述符,专注于已批准药物的化学空间定位与对比分析。
- • 主要局限:仅支持PCA降维;单次分析上限为1,000个化合物,不适用于大规模筛选库。
ChemGPS
- • 定位:快速理化性质分析的化学空间导航工具,适合需要快速查看化合物理化空间分布的场景,推荐与MAYA互补使用(ChemGPS做快速侦察,MAYA做深度分析)。
4.2 相似度预测工具
相似度预测的核心任务是基于化合物的结构特征,预测其潜在的靶标相互作用谱——这是靶标去卷积(Target Deconvolution)和老药新用研究的基础方法。
3DSTarPred
- • 获取地址:3dstarpred.pumc.wecomput.com
- • 核心方法:基于三维构象相似性(3D形状比对)预测靶标,整合ADMET性质评估与完整计算工作流。
- • 性能基准测试(118个FDA批准药物):
工具 | 预测方法 | 准确率 |
|---|---|---|
3DSTarPred | 3D构象相似性 | 76.3% |
SwissTargetPrediction | 2D/3D相似性 | 72.9% |
NetInfer | 网络推断 | — |
SEA | 化学相似性集成 | — |
FastTargetPred | 快速靶标预测 | — |
- • 处理速度:约1,000化合物/分钟(3D相似性搜索)
- • 主要局限:
- • 依赖远程服务器,大规模库(>100,000化合物)因计算需求受限;
- • 对训练数据中未覆盖的新骨架,假阳性率偏高;
- • 预测结果依赖ChEMBL等公共数据库的覆盖度与标注质量。
SwissTargetPrediction
- • 采用2D/3D双模态相似性进行靶标预测,是目前学术界最常用的在线靶标预测工具之一,可作为3DSTarPred的互补验证工具。
4.3 定量分析工具(QSAR / multi-QSAR)
定量分析框架的本质是建立分子描述符与活性/性质之间的数学映射,QSAR(Quantitative Structure-Activity Relationship)是其经典形式。
QSAR 到 multi-QSAR 的演进
传统 QSAR
→ 单一活性端点
→ 线性/非线性描述符-活性映射
→ 局限:每个靶标需独立建模,无法捕获靶标间相关性
多维 QSAR(multi-QSAR / Deep QSAR)
→ 多活性端点同步建模
→ 整合 AI/深度学习方法
→ 支持药效动力学 + ADMET 参数联合优化
→ 能够量化靶标选择性与多靶点活性权衡SARANEA
- • 获取地址:lifescienceinformatics.uni-bonn.de
- • 核心功能:
- • 可视化网络状相似图(Network-like Similarity Graphs)
- • 同时分析化合物相似度与效价/选择性
- • 活性悬崖识别与可视化
- • 友好的图形用户界面
- • 主要局限:不支持多种表示方式的同步交叉分析;仅支持指纹(fingerprint)类描述符,不支持连续型描述符。
DTINet
- • 代码仓库:github.com/luoyunan/DTINet
- • 核心方法:通过异质信息网络整合多源数据(蛋白质-蛋白质相互作用网络、药物相似性、疾病关联等),预测药物-靶标相互作用(DTI),并支持对无已知靶标的新化合物进行预测。
- • 多靶点应用:特别适合药物重定向场景,可挖掘已批准药物在新靶标上的潜力。
- • 主要局限:对高度研究靶标存在数据偏倚;不预测定量亲和力(仅给出二元预测)。
4.4 机器学习与深度学习工具
机器学习已成为现代药物发现流程中不可或缺的基础设施,其在 SMARTs 分析中的核心优势在于:从高维化学/生物数据中自动提取非线性关联,实现多分子特征的同步优化。
Chemprop(v2)
- • 代码仓库:github.com/chemprop/chemprop
- • 核心架构:D-MPNN(Direct Message Passing Neural Networks,直接消息传递神经网络)
- • 直接在分子图结构上进行特征传播,无需预定义描述符;
- • 原子级消息传递机制,捕获局部化学环境;
- • 支持边(化学键)信息的显式编码。
- • 主要功能:
- • 分子性质多任务预测(分类 + 回归)
- • 原子映射反应分析
- • 超参数自动优化
- • 红外光谱分析
- • 用户自定义特征融合
- • SMARTs 适用性:多任务学习框架天然支持多靶点活性同步建模。
- • 主要局限:不直接整合三维构象信息。
Polygon
- • 代码仓库:github.com/bpmunson/polygon
- • 核心方法:生成式强化学习(Generative Reinforcement Learning),专为从头(de novo)设计多药理学化合物而构建。
- • 验证结果:
- • 在含109,811个化合物(两蛋白靶标实验活性数据)的验证集上,分类准确率达 81.9%;
- • 32个 de novo 设计化合物经合成后针对 MEK1 和 mTOR 进行实验验证,多数表现出 >50% 抑制率;
- • 在同一数据集内对双靶标活性化合物的分类保持稳定性能。
- • 工作流建议:与 Chemprop 协同使用——Polygon 生成候选结构 → Chemprop 快速评估多维性质 → 迭代反馈驱动设计。
- • 主要局限:数据稀缺性对生成质量影响显著;模型可解释性不足。
OpenChem
- • 代码仓库:github.com/Mariewelt/OpenChem
- • 定位:面向计算化学与药物设计的深度学习工具库,提供模块化的模型构建接口。
- • 支持模型类型:分类、回归、生成模型
- • 在案例研究中的应用:预测抗菌肽的两性性指数(Amphiphilicity Index),R²=0.68(详见第7节)。
- • 主要局限:缺乏超参数自动优化支持;无GPU时训练速度极慢,实用性受限。
DeepMol
- • 代码仓库:github.com/BioSystemsUM/DeepMol
- • 核心特色:
- • 自动化机器学习与深度学习框架,一体化端到端流程;
- • 内置多种分子标准化方案;
- • 多特征提取方法(指纹、描述符、图神经网络);
- • 特征重要性分析(可解释性支持);
- • 模块化设计,易于扩展。
- • 主要局限:同样受限于GPU依赖,无GPU环境下大规模任务不切实际。
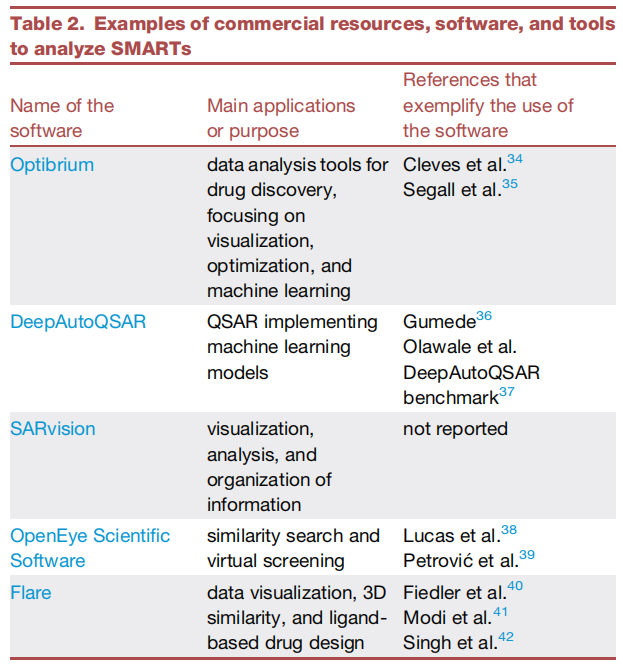
5. 商业工具概览
综述同时评估了5个商业平台,对于拥有预算的学术课题组或工业界团队,其在可用性、技术支持与更新频率上具有优势:
软件 | 主要功能 | 多靶点特色 | 可及性 |
|---|---|---|---|
Optibrium (StarDrop/BioPharmics) | 数据分析、SAR可视化、ADMET-QSAR、多参数优化 | 自定义交互图形;3D信息整合;MPO打分 | 商业授权(提供学术/非营利许可) |
DeepAutoQSAR | QSAR建模(Schrödinger生态) | 自动化机器学习模型构建 | 商业订阅 |
SARvision | SAR信息可视化与组织 | 大数据集交互分析 | 商业授权 |
OpenEye Scientific | 相似度搜索、虚拟筛选 | 基于形状(ROCS)与药效团的3D搜索 | 商业授权(提供学术许可) |
Flare | 数据可视化、3D相似度、配体药物设计 | FEP+/RBFE准确性;多靶点结合模式分析 | 商业授权 |
开源 vs. 商业工具核心权衡
开源工具 商业工具
+ 零成本获取 + 技术支持与培训
+ 透明可审计 + 持续更新维护
+ 科研定制灵活 + 优化用户体验
+ 社区共建 + 更广功能覆盖
- 文档质量参差不齐 - 高昂许可费用
- 长期维护依赖科研经费 - 源代码不透明
- 非专业用户上手门槛高 - 定制化能力受限6. 工具选择矩阵
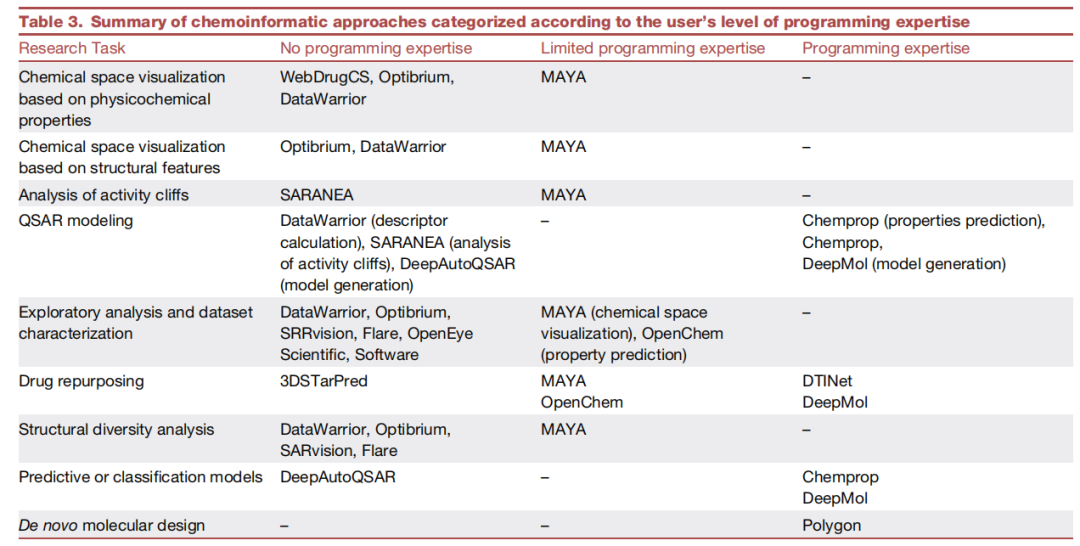
综述提供了一份依据用户编程能力分层的工具推荐矩阵,这是本文最具实践价值的贡献之一:
研究任务 | 无编程基础 | 有限编程能力 | 编程专家 |
|---|---|---|---|
基于理化性质的化学空间可视化 | WebDrugCS, Optibrium, DataWarrior | MAYA | — |
基于结构特征的化学空间可视化 | Optibrium, DataWarrior | MAYA | — |
活性悬崖分析 | SARANEA | MAYA | — |
QSAR建模 | DataWarrior(描述符计算), SARANEA(活性悬崖), DeepAutoQSAR | — | Chemprop, DeepMol |
探索性分析与数据集表征 | DataWarrior, Optibrium, SARvision, Flare, OpenEye | MAYA, OpenChem | — |
药物重定向 | 3DSTarPred | MAYA, OpenChem | DTINet, DeepMol |
结构多样性分析 | DataWarrior, Optibrium, SARvision, Flare | MAYA | — |
预测/分类模型构建 | DeepAutoQSAR | — | Chemprop, DeepMol |
从头分子设计(De novo) | — | — | Polygon |
7. 实战案例:短肽多靶点设计中的 SMARTs 集成工作流
7.1 短肽作为多靶点药物支架的优势
短肽(<50个氨基酸)正在成为多靶点药物开发的重要支架类别。与传统小分子相比,经合理设计的短肽具有:
- • 更高的靶标选择性(通过序列工程精细调控)
- • 更低的系统毒性(可生物降解)
- • 广泛的治疗潜力(抗菌、抗病毒、抗肿瘤、免疫调节)
- • 可利用 AI/ML 实现快速序列空间探索
APEXDUO 是一个典型案例——这是一个专为预测具有多模态性质肽段而设计的 AI 驱动模型,代表了将肽设计整合进化学信息学工作流的新兴趋势。
7.2 工作流详解:OpenChem + MAYA 集成管线
第一步:数据准备
- • 数据来源:DBAASP(Database of Antimicrobial Activity and Structure of Peptides, v3)
- • 筛选条件:
- • 单体肽
- • 序列长度 < 50 个氨基酸
- • 针对WHO耐药优先级病原体(A. baumannii、P. aeruginosa、E. faecium、S. aureus 等)具有实验验证活性
- • 最终数据集:2,551条肽序列(训练/验证)
- • 分子表示:序列 → SMILES字符串(用于指纹计算与神经网络输入)
第二步:OpenChem 回归模型训练
预测目标为两性性指数(Amphiphilicity Index)——抗菌肽穿透细菌膜的关键物理化学参数,反映肽段疏水/亲水区域的空间分布。
网络架构:
输入层(SMILES编码)
↓
全连接层 1: 1024个神经元 + Dropout(0.5)
↓
全连接层 2: 512个神经元 + Dropout(0.5)
↓
全连接层 3: 128个神经元 + Dropout(0.5)
↓
输出层: 1个神经元(两性性指数回归值)训练配置:
- • 数据集划分:80% 训练 / 20% 验证
- • 优化器:Adam(gamma=0.9)
- • 评估指标:外部验证 R²
性能结果:
指标 | 值 |
|---|---|
外部验证 R² | 0.68 |
外部测试集平均 RMSD | 0.22(35肽 vs. DBAASP服务器预测值) |
误差分析:高 RMSD 主要集中于训练集中结构基序欠代表的肽段,提示线性 SMILES 表示无法充分捕获三维构象特征——这是肽段相关三维描述符整合的重要方向。
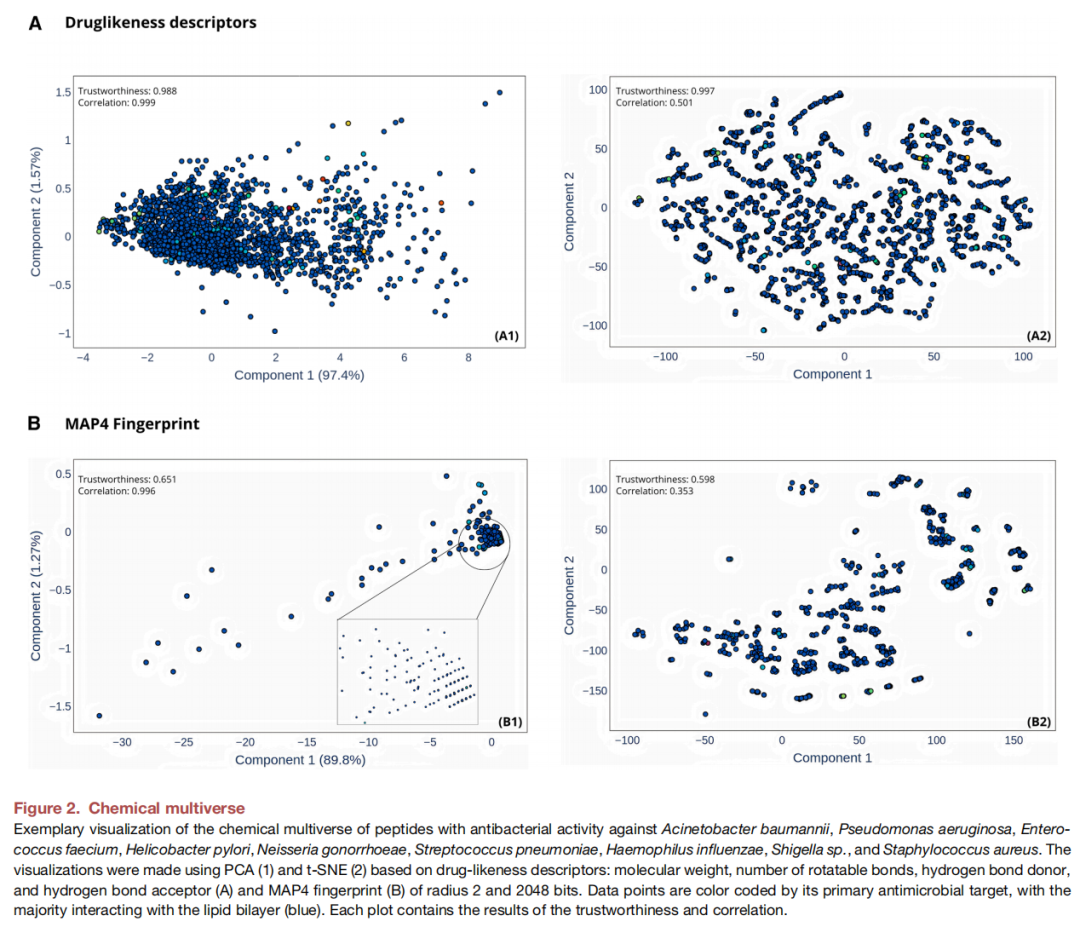
第三步:MAYA 化学多元宇宙可视化
分析对象:2,570个化合物(包含上述抗菌肽数据集)
双描述符体系:
描述符类型 | 内容 | 降维方法 | 保留信息 |
|---|---|---|---|
理化(药物样)描述符 | 分子量、可旋转键数、氢键供体/受体 | PCA + t-SNE | 全局理化相似性 |
MAP4指纹(半径2, 2048 bit) | 基于最小哈希+SMILES的拓扑指纹 | PCA + t-SNE | 局部结构特征 |
关键发现:
- 1. 基于理化描述符的PCA/t-SNE图(A1/A2):不同抗菌靶标的肽广泛分散于化学空间,无明显聚类——说明单纯靠理化性质无法区分不同靶标的抗菌肽,符合一般药物样描述符不编码靶标特异性信息的预期;
- 2. 基于MAP4指纹的PCA/t-SNE图(B1/B2):展现出更强的局部聚类趋势——尽管不同靶标的肽在整体理化空间上高度重叠,其序列/结构特征仍可被指纹表示区分。这表明结构指纹在识别靶标相关结构基序方面优于理化描述符。
第四步:整合决策
通过将 OpenChem 预测的两性性指数与 MAYA 生成的化学空间分布相结合:
- • 识别同时具有高两性性(有利于膜穿透)与特异性结构聚类(特征结构基序)的候选肽段;
- • 定位对脂质双层和外排泵具有双重作用潜力的多靶点候选物;
- • 为后续实验验证提供优先级排序依据。
8. 现存局限与关键挑战
8.1 数据层面
挑战 | 具体表现 | 潜在影响 |
|---|---|---|
数据集不平衡 | 热门靶标数据量远超罕见靶标 | 预测模型对欠代表靶标产生系统性偏差 |
阴性/非活性数据稀缺 | 公共数据库以活性数据为主 | 模型边界不清,假阳性率偏高 |
实验数据标准化不足 | 不同来源的检测格式、活性阈值、标注规范差异显著 | 相似度预测可靠性降低 |
多靶点基准数据集缺失 | 无社区公认的多靶点标准评估集 | 方法间客观比较困难,模型鲁棒性难以评估 |
8.2 方法层面
- • 三维信息整合不足:多数开源工具仍依赖二维描述符/指纹,忽略构象特征——对肽类化合物尤为不利;
- • 深度模型可解释性差:神经网络的"黑箱"特性限制了化学家对结果的机理理解;
- • 域适用性(Domain of Applicability)界定模糊:大多数工具缺乏明确的预测适用范围声明,用户难以评估特定化合物的预测可信度;
- • 多靶点验证标准缺失:SMARTs 预测结果的实验验证策略尚无共识,限制了计算模型向临床决策的转化。
8.3 工具生态层面
- • 开源工具长期维护困难:大多数开源工具依托学术课题组,经费不确定性导致更新停滞、文档缺失;
- • 转化鸿沟持续存在:in silico SMARTs 预测与细胞/动物/临床结果之间的相关性往往较弱或高度情境依赖;
- • 有益多药理学 vs. 滥靶性的区分难题:需要结合安全性、选择性与治疗情境综合判断,而非单纯依赖统计关联。
9. 未来展望
综述提出了以下高优先级研究方向:
9.1 数据与基准建设
- • 建立标准化、多靶点、跨机构的基准数据集
- • 系统纳入高质量阴性数据(inactive compounds)
- • 整合临床标注信息,推动 in silico-clinical 转化研究
9.2 方法论创新
- • 三维描述符集成:尽管构象灵活性与集成计算成本仍是挑战,但3D描述符(尤其用于肽类和大环化合物)代表了重要发展方向;
- • 多组学数据整合:将转录组、蛋白组、代谢组数据纳入 SMARTs 分析框架;
- • 可解释 AI(XAI):发展可为化学家提供机理洞见的可解释深度学习方法;
- • 混合治疗学:将分析范围从小分子延伸至肽-小分子杂合物、抗体偶联药物等。
9.3 生态系统建设
- • 建立开源工具协作维护机制(类似 OpenBabel、RDKit 的社区模式)
- • 推动社区验证与方法论精化的良性反馈循环
- • 开发更统一的 API 接口,降低工具间集成门槛
写在最后
值得注意的是,文章坦承 in silico 预测与临床结果之间的弱相关性问题——这恰恰是该领域长期存在的"可重复性危机"的延伸。SMARTs 工具箱的真正价值,在于为实验验证提供有据可查的优先级假设,而非取代湿实验判断。随着集成多组学数据的下一代工具涌现,这一转化鸿沟有望逐步缩小。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-28,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号