J. Med. Chem. | 成功药物优化的规律到底是什么?

J. Med. Chem. | 成功药物优化的规律到底是什么?

DrugIntel
发布于 2026-04-21 11:13:15
发布于 2026-04-21 11:13:15

来源:What Happens in Successful Optimizations? A Survey of 2018–2024 Literature, Paul D. Leeson, Journal of Medicinal Chemistry 2026 69 (6), 6337-6395 作者:Paul D. Leeson(Paul Leeson Consulting Ltd) DOI:10.1021/acs.jmedchem.5c03171
一、研究背景与意义
从苗头化合物(hit)到临床候选药物(candidate)的优化过程,是药物化学的核心实践。然而,长期以来,药物化学界对"成功优化究竟发生了什么"的认识,主要依赖小规模数据集或单一靶标类别的经验积累,缺乏系统性的定量分析。
本文作者 Paul Leeson 自2018年起,历时数年,从2018—2024年的药物化学文献中手工阅读并筛选,最终收集了 487对起始化合物→候选药物的优化对(start-to-candidate pairs),涵盖498个候选药物,是迄今为止从公开文献中构建的最大规模此类数据集。
分析维度涵盖:靶标类别、疾病领域、起始化合物来源、作用机制、给药途径,以及超过30项分子理化性质的系统性变化规律。
值得注意的是,由于文献发表滞后,数据集所代表的实际创新高峰期为2011—2019年(专利优先权日中位年份为2014年),平均从专利申请到文献发表间隔约7年。
二、数据集构成
2.1 文献来源
- • Journal of Medicinal Chemistry(JMC)贡献最多,占68%
- • ACS Medicinal Chemistry Letters(11%)、Bioorganic & Medicinal Chemistry Letters(7%)等补充
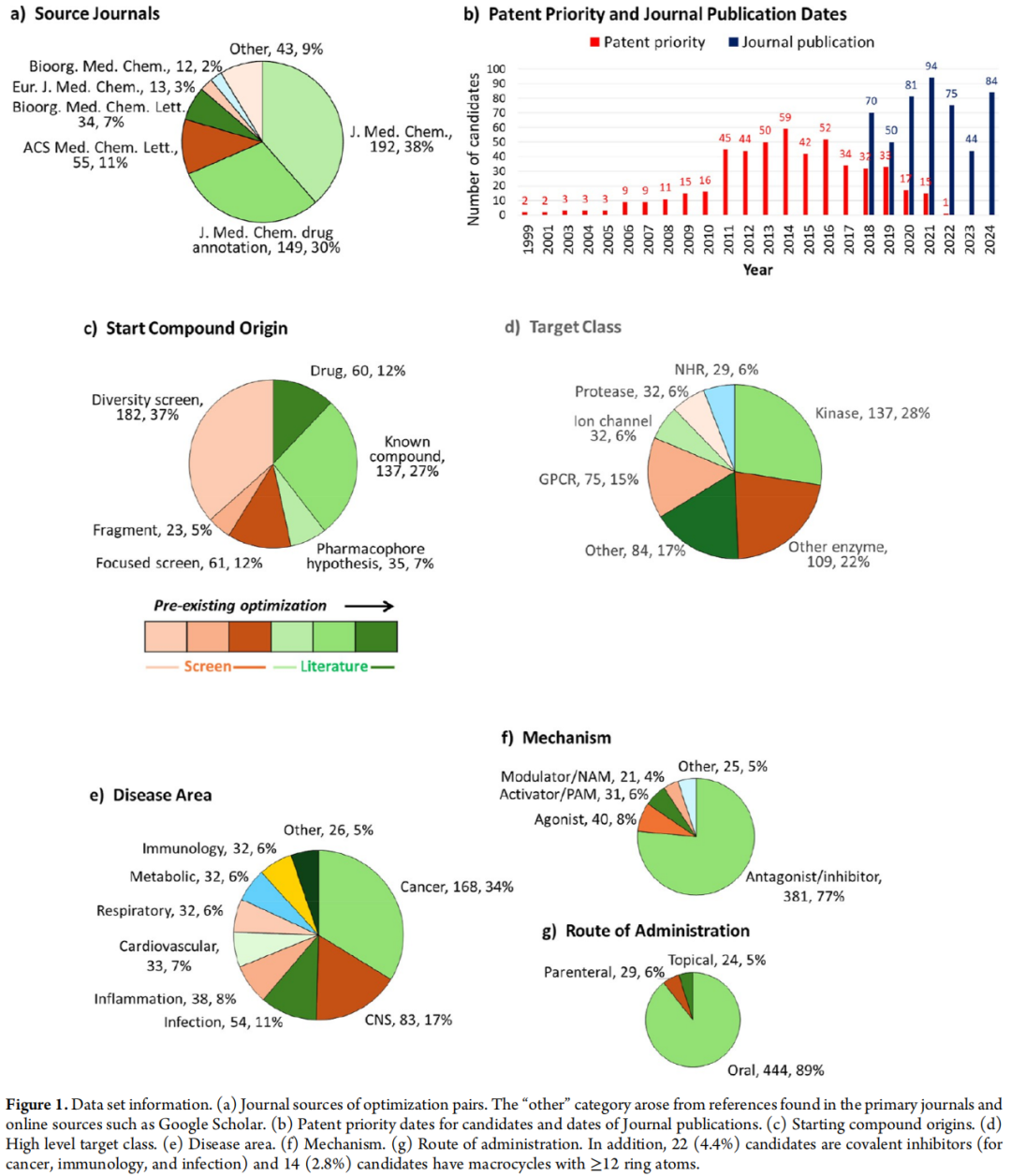
2.2 起始化合物来源分类
来源类型 | 占比 | 说明 |
|---|---|---|
多样性筛选(HTS / DEL / 表型筛选) | 37% | 最主要来源,远高于前人报道的21% |
已知文献化合物 | 27% | 预优化程度不等 |
已上市药物 | 12% | 内置优化属性 |
聚焦筛选(虚拟筛选 / SBDD / 配体筛选) | 12% | 结合靶标信息 |
药效团假说设计 | 7% | 主观性较强 |
片段筛选(FBDD) | 5% | 低分子量,高增长空间 |
方法论差异说明: 本研究相较于Brown(2023)等前期工作,将来自多样性筛选的比例从21%修正至37%,原因在于本研究追溯了"已知化合物"的原始筛选来源,避免了来源误判。
2.3 靶标类别分布
激酶(28%)> 其他酶(22%)> GPCR(15%)> 蛋白酶(6%)> 离子通道(6%)= 核激素受体(6%)
2.4 疾病领域与给药途径
- • 肿瘤学(34%)占主导,其次为CNS(17%)和感染性疾病(11%)
- • 口服给药占89%,86.3%的口服候选药物符合 Lipinski Ro5
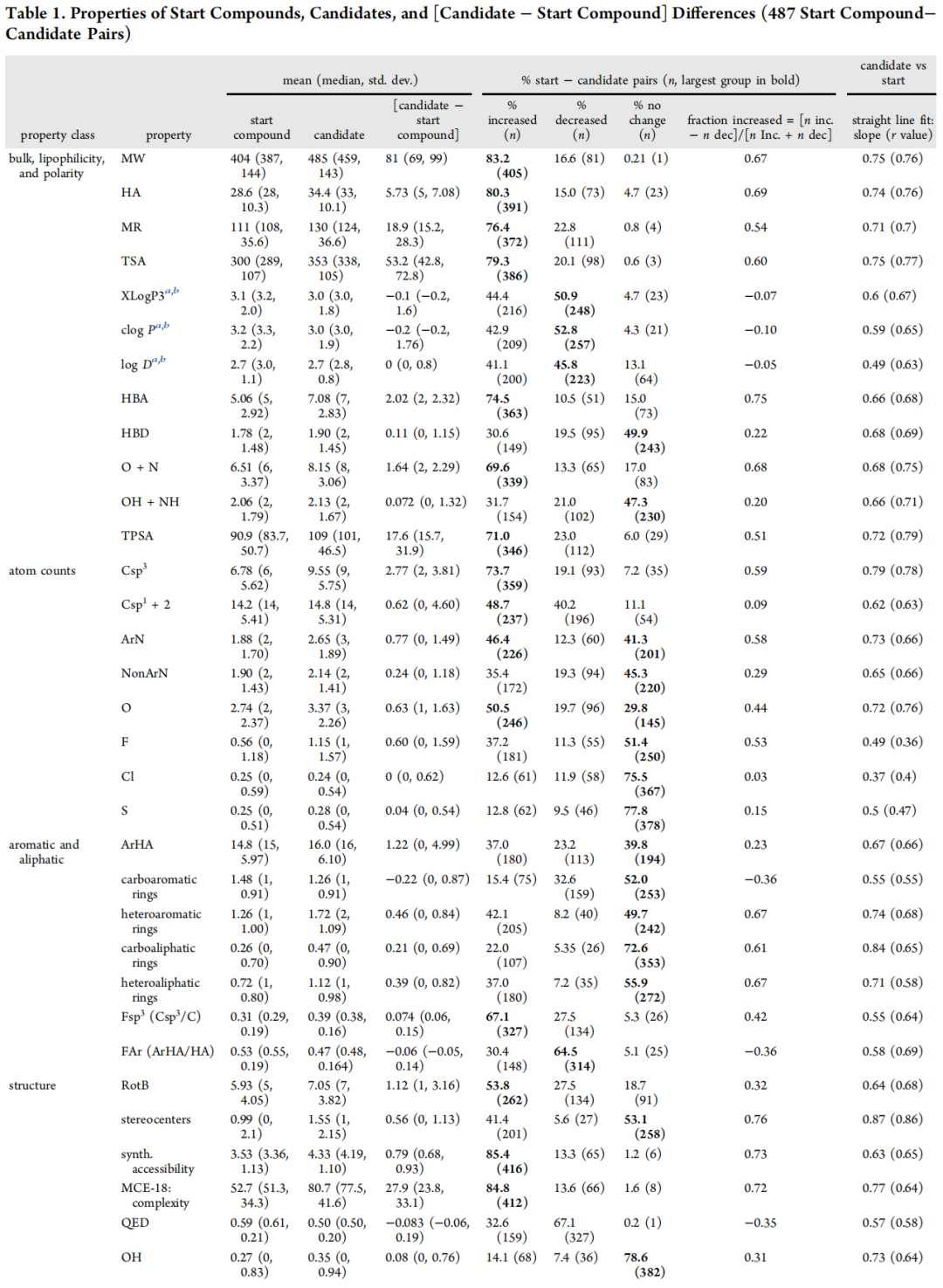
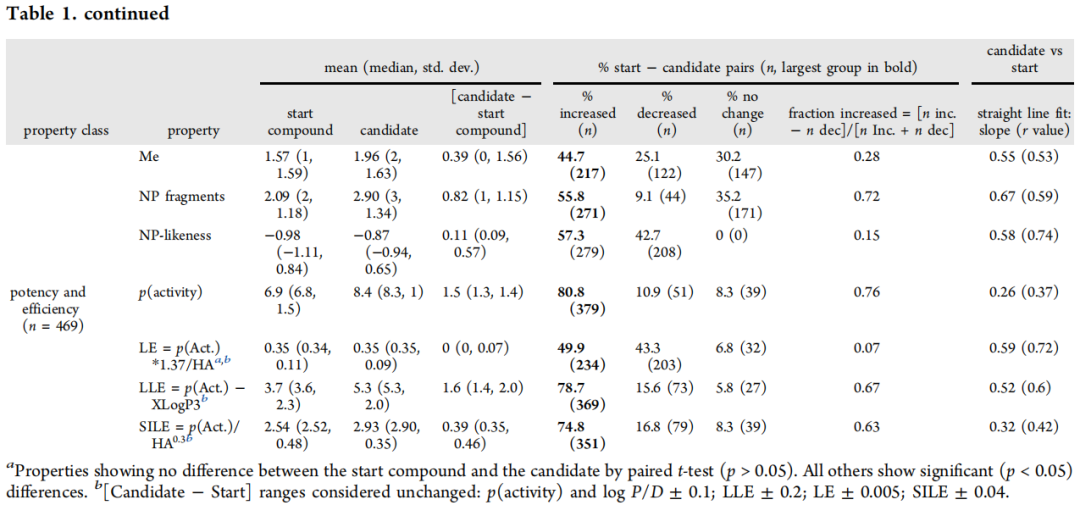
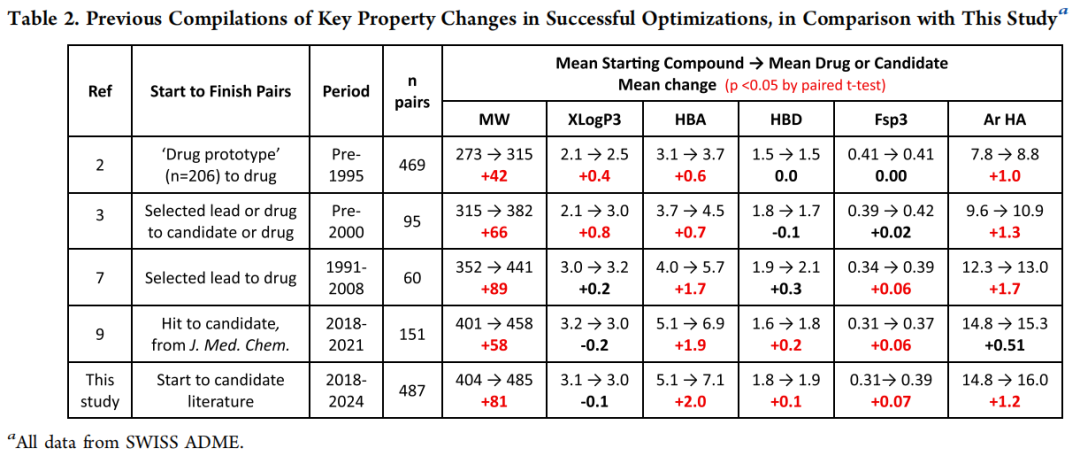
三、整体理化性质变化规律
3.1 关键性质变化汇总
性质 | 起始化合物(均值) | 候选药物(均值) | 变化量 | 增加概率 |
|---|---|---|---|---|
分子量 MW(Da) | 404 | 485 | +81 | 83.2% |
重原子数 HA | 28.6 | 34.4 | +5.73 | 80.3% |
XLogP3 | 3.1 | 3.0 | -0.1(无显著变化) | 44.4% |
cLogP | 3.2 | 3.0 | -0.2 | 42.9% |
HBA | 5.06 | 7.08 | +2.02 | 74.5% |
HBD | 1.78 | 1.90 | +0.11(无显著变化) | 30.6% |
TPSA(Ų) | 90.9 | 109 | +17.6 | 71.0% |
Csp³ 原子数 | 6.78 | 9.55 | +2.77 | 73.7% |
芳香氮原子数 ArN | 1.88 | 2.65 | +0.77 | 46.4% |
F 原子数 | 0.56 | 1.15 | +0.60 | 37.2% |
杂芳香环数 | 1.26 | 1.72 | +0.46 | 42.1% |
碳芳香环数 | 1.48 | 1.26 | -0.22 | 15.4%(减少) |
手性中心数 | 0.99 | 1.55 | +0.56 | 41.4% |
MCE-18(复杂度) | 52.7 | 80.7 | +27.9 | 84.8% |
p(activity) | 6.9 | 8.4 | +1.5 | 80.8% |
LLE(LipE) | 3.7 | 5.3 | +1.6 | 78.7% |
LE | 0.35 | 0.35 | 0(无显著变化) | 49.9% |
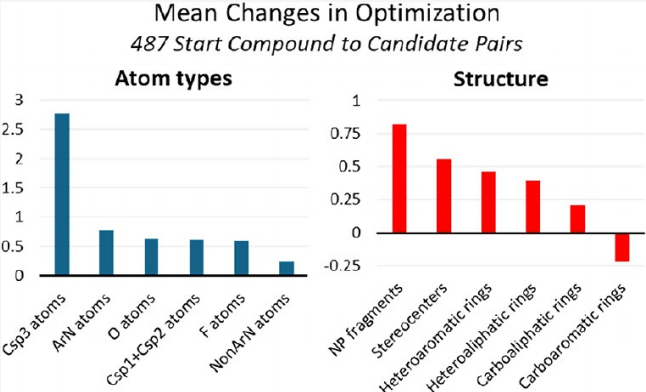
3.2 核心规律解读
① 分子量增长是药物优化的"内在驱动力"
MW平均增长81 Da,与体外活性(p(activity))平均提升1.5个单位强相关,反映了增大范德华接触面积对结合亲和力的贡献。从不同起始来源看,"MW距离"与其所需的活性提升幅度成正比:片段筛选苗头(+191 Da)> 多样性筛选(+76 Da)> 已知化合物(+40 Da)> 已上市药物(+27 Da)。
② 亲脂性的精准控制——优化的最难之处
平均亲脂性几乎不变(ΔXLogP3 = -0.1),但这一平均值掩盖了大量个体变化:约50.9%的案例亲脂性增加,44.4%降低。片段筛选起点是唯一平均亲脂性显著增加的类别(+1.4),而多样性筛选起点的亲脂性则平均降低(-0.5)。这说明在分子量增长的同时维持亲脂性稳定,是药物化学家主动为之的结果,而非自然发生。
③ HBA显著增加,HBD几乎不变
HBA平均增加2.02,而HBD仅增加0.11(无统计学显著性)。这一不对称性反映了一条清晰的策略:通过增加极性H键受体(而非供体)来控制亲脂性,同时避免HBD增加对膜通透性的负面影响。HBA/HBD比值随时间持续增大,是当代候选药物的重要趋势。
④ 配体效率(LE)不变,LLE显著提升
LE(= p(activity)×1.37/HA)在整体数据集中均值不变,是因为HA和活性同步增加所致。但LLE(= p(activity) − XLogP3)平均增加1.6,显示在亲脂性控制下活性的净提升,是衡量优化质量更敏感的指标。仅在多样性筛选亚组中,LE有小但显著的提升(+0.02, p=0.0012)。
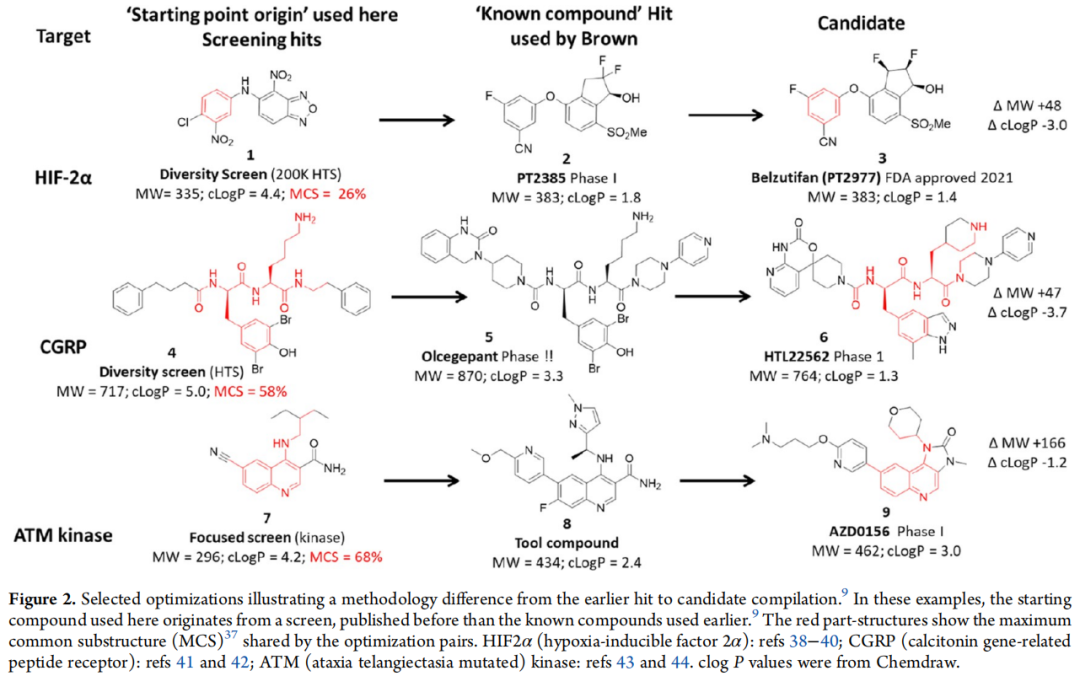
四、分子变化的深层来源分析
4.1 重原子增长的原子类型分解
HA平均增加5.73,其来源按贡献排序:
以上六类原子合计贡献了HA增量的98%。
4.2 归一化性质变化分析(排除HA增长的稀释效应)
通过将各性质除以HA数进行归一化,识别出真正超比例增长的性质(即在候选药物中密度显著提升的性质):
超比例增长(>50%优化对增加,且归一化值显著提升):
- • MCE-18(复杂度):最显著的单一影响因素
- • Csp³原子密度:+4.6 per 100 HA
- • HBA密度
- • NP片段数
超比例增长但增加概率<50%(高影响但非普遍):
- • F原子、手性中心、脂肪环(碳环和杂环)、杂芳香环、ArN原子
与HA等比例增长(密度不变):
- • 合成可及性、可旋转键数、TPSA、O原子数、非芳香N原子数
低于HA比例增长(密度下降):
- • HBD、碳芳香环(苯环)、Csp²+Csp¹原子
4.3 脂肪复杂性的三维代理指标
以手性中心、杂脂肪环、碳脂肪环三个参数的联合变化作为"脂肪复杂性"的代理指标:
- • 三者均无变化:23%
- • 至少一个增加,其余不变:55.9%(最主要模式)
- • 至少一个增加且至少一个减少:7.8%
- • 至少一个减少,其余不变:8%
这一分析清晰地表明,引入脂肪复杂性是现代药物优化的主旋律,而非例外。
五、芳香环系统的演变规律
5.1 苯环减少,杂芳香环增加
碳芳香环(主要为苯环)平均减少0.22个,而杂芳香环平均增加0.46个。这一反向趋势在所有靶标类别和起始来源中均保持一致,是最具普遍性的结构优化规律之一。
其背后的化学逻辑包括:
- • 杂芳香环引入氮原子,降低亲脂性("必要的氮原子"效应)
- • 苯环含量高的分子通常具有较高的平面性,不利于溶解度
- • 杂芳香环可提供更丰富的氢键受体机会
- • 芳香碳环与更高的开发风险相关(与已上市药物相比,其他靶标化合物中苯环含量更高)
5.2 常见环系统分析
487对优化数据中,前46种环系统(出现≥5次)覆盖了全部环系统的77.1%。在候选药物中出现频率显著高于起始化合物的环系统包括:
吡啶 > 环丙烷 > 吡唑 > 吡咯烷 > 吗啉 > 嘧啶 > 吡喃 > 噁丁环(oxetane)
其中噁丁环在起始化合物中完全未出现,但在11个候选药物中出现。这些环系统同时也是天然产物片段,印证了NP骨架的核心地位。
5.3 激酶类靶标的特异性
激酶起始化合物具有最高的杂芳香环密度,但在优化过程中杂芳香环密度下降,与其他靶标类别(均为增加)相反。这是因为激酶苗头化合物通常富含杂环(针对ATP结合口袋的已有认知),优化过程反而需要引入更多脂肪性成分来改善选择性。
六、片段筛选(FBDD)的特殊规律
6.1 FBDD候选药物的结构特征
与非FBDD候选药物相比,FBDD候选药物具有:
- • 更高的芳香原子比例(FAr: 0.54 vs 0.47)
- • 更低的Fsp³(0.32 vs 0.39)
- • 更高的芳香环数(平均3.48 vs 2.96)
这一特征在本研究(n=23)中与文献报道的54个FBDD临床化合物高度一致。
6.2 FBDD优化轨迹的独特性
FBDD起始化合物在碳芳香环和杂芳香环密度上几乎无变化,这与其他策略(起始化合物芳香环密度显著变化)形成对比。这可能反映了:一方面片段苗头本身已富含芳香性;另一方面,FBDD的增长策略倾向于在保留核心片段的基础上进行三维延伸。
七、天然产物相关性分析
7.1 NP片段的核心地位
研究使用Over等(2013)定义的NP片段库,分析了每个优化对中NP片段数的变化:
- • 起始化合物平均NP片段数:2.09
- • 候选药物平均NP片段数:2.90(增加0.82,p<0.05)
- • 十大最常用NP片段:吡啶、哌啶、吡咯烷、吡唑、环丙烷、哒嗪、吗啉、咪唑、嘧啶、环己烷
7.2 伪天然产物(PNP)的主导地位
基于Greiner等(2025)的PNP分类:
- • 候选药物中PNP占比:66%(起始化合物为46%)
- • 候选药物成为PNP的概率是起始化合物的2.3倍(OR=2.3, 95%CI: 1.8–3.0, p<0.0001)
- • 在所有靶标类别和起始来源中,PNP比例在候选药物中均高于起始化合物(已上市药物起点除外)
这与临床化合物整体趋势吻合:PNP占比从1990年代的30%上升至2010年后的67%,反映了NP骨架在现代药物设计中的深远影响。
八、最大公共子结构(MCS)分析
8.1 起始结构的保留程度
- • 全数据集MCS均值:70.0%(中位数70.6%,标准差21.5%)
- • 随机打乱配对的对照组MCS均值仅34.4%,差异极显著
- • 46对(9.4%)实现MCS=100%,即候选药物完全保留起始结构骨架
8.2 不同来源的MCS比较
起始来源 | MCS均值(%) |
|---|---|
已上市药物 | 77.6 |
已知化合物 | 71.2 |
药效团假说 | 78.0 |
聚焦筛选 | 70.6 |
片段筛选 | 75.0 |
多样性筛选 | 63.9(最低) |
多样性筛选起点MCS最低,与其起始化合物质量最参差不齐、需要更大改造幅度相符。
8.3 实践意义
MCS高达70%意味着:选对起始化合物,本身就决定了候选药物结构的大半。在Hit-to-Lead阶段尽早确定核心骨架(preferred scaffold),是提高优化效率的关键策略。
九、规则五(Ro5)合规性分析
9.1 总体合规情况
487个口服候选药物中(n=445),86.3%符合Ro5(违反0或1条规则),与Brown 2018–2021数据高度一致。
9.2 违规分布不均衡
Ro5参数 | 违规比例 |
|---|---|
MW > 500 | 30.6%(最主要违规来源) |
cLogP > 5 | 14.0% |
O+N > 10 | 8.1% |
OH+NH > 5 | 0.9%(极少违规) |
数据表明:当前候选药物的分布已不符合经典Ro5的"5"的助记规则——MW和cLogP的90百分位分别为586 Da和5.4,超过500/5的截止值;而OH+NH截止值5明显偏高。
9.3 bRo5候选药物
采用Doak等提出的扩展分类(eRo5: MW 500–700; bRo5: MW>700或其他参数超出):
- • eRo5候选药物:102个(22.9%)
- • bRo5候选药物:34个(7.6%)
bRo5分子(Protac、大环、分子胶水等)通常依赖分子变色龙性(chameleonicity)——在非极性环境中通过分子内氢键遮蔽极性表面,实现出乎意料的口服吸收。实验性极性表面积(EPSA)是评估这一特性的重要工具。
十、前沿优化案例精析
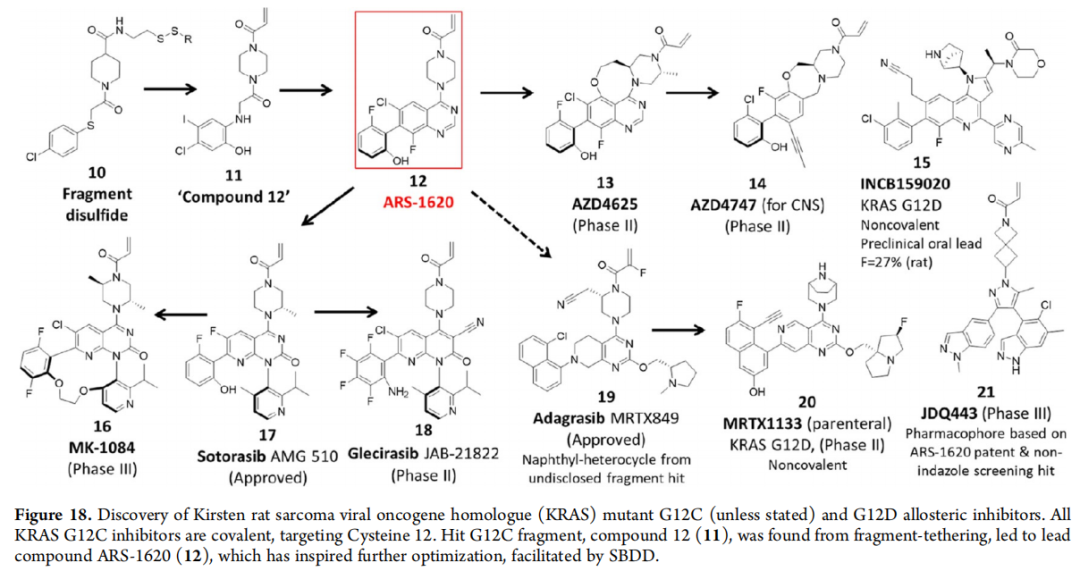
10.1 KRAS抑制剂——"跟风效应"的典型案例
从2013年Shokat课题组发现G12C共价抑制剂(fragment tethering,化合物11)到ARS-1620(12),再到:
- • Sotorasib(17):首个获批KRAS G12C药物,引入吡啶环填充cryptic pocket
- • Adagrasib(19):萘基来自未公开筛选苗头,胺侧链增加多样性
- • MRTX1133(20):非共价G12D抑制剂,桥式哌嗪靶向Asp-12
- • KRAS三元复合物大环系列(166–169):以亲环素A配体sanglifehrin A(164)为起点,通过二硫键拴系策略确定大环化策略
KRAS系列候选药物的平均理化性质(MW 586, XLogP3 4.9, MCE-18 125)显著高于ARS-1620(MW 431, XLogP3 4.0, MCE-18 56),但亲脂性控制良好,体现整体趋势。
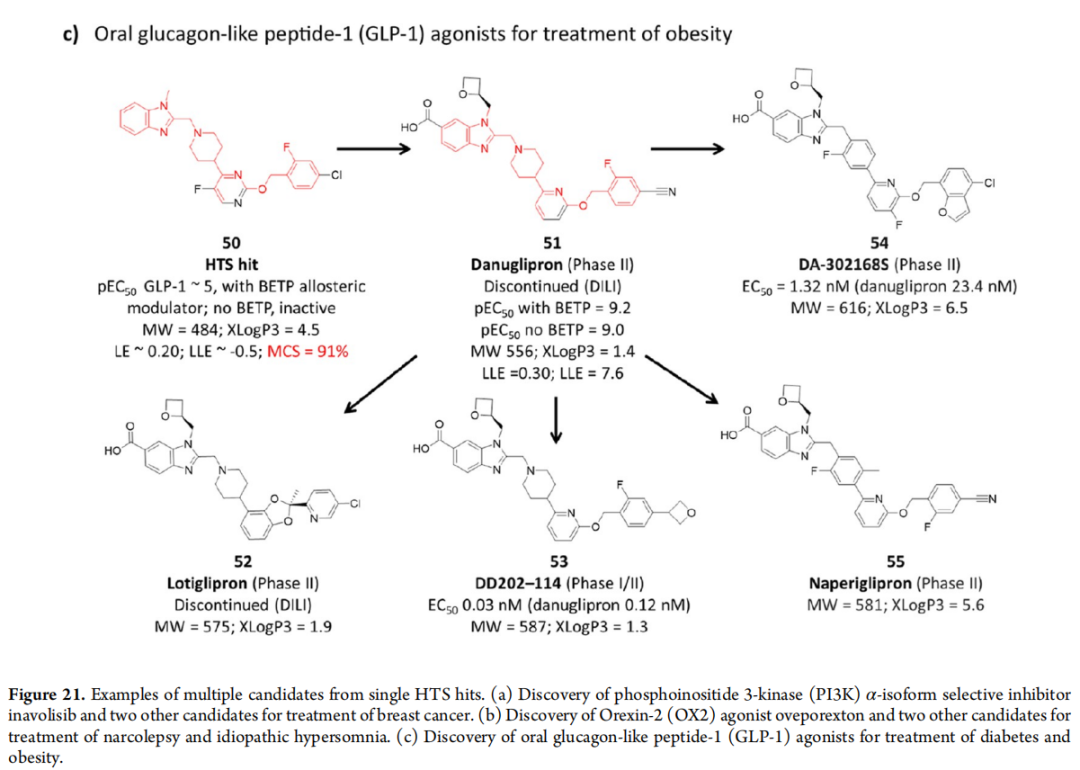
10.2 口服GLP-1受体激动剂
Danuglipron(51)来自一个仅在BETP激活的GLP-1蛋白上有微弱活性的HTS苗头(50),但MCS高达91%——优化几乎保留了全部起始骨架,核心改变是在苯并咪唑5位引入羧基模拟GLP-1底物的酸性残基。该案例是"高质量苗头→高MCS优化"的绝佳示范。
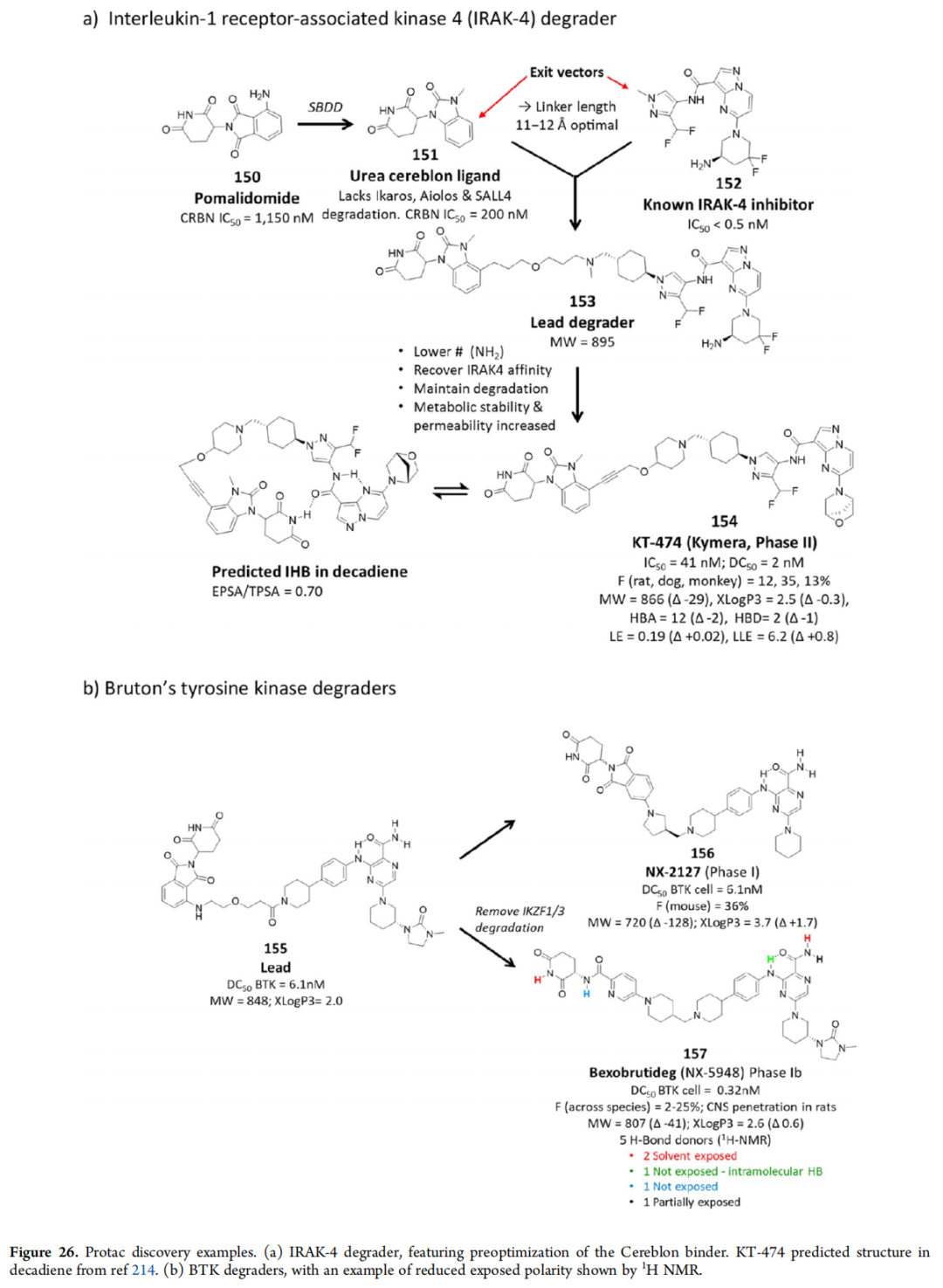
10.3 Protac案例——KT-474与NX-2127
KT-474(IRAK-4降解剂)的关键策略:
- 1. 预优化Cereblon E3连接酶配体(151),避免降解相关转录因子
- 2. 晶体学确定退出向量(exit vectors)
- 3. 连接子长度优化(13原子最优)
- 4. 同步优化IRAK-4结合端(152)和连接子
最终候选药物154(MW 866)展示分子内氢键,EPSA/TPSA = 0.70,是大分子口服吸收的关键特征。
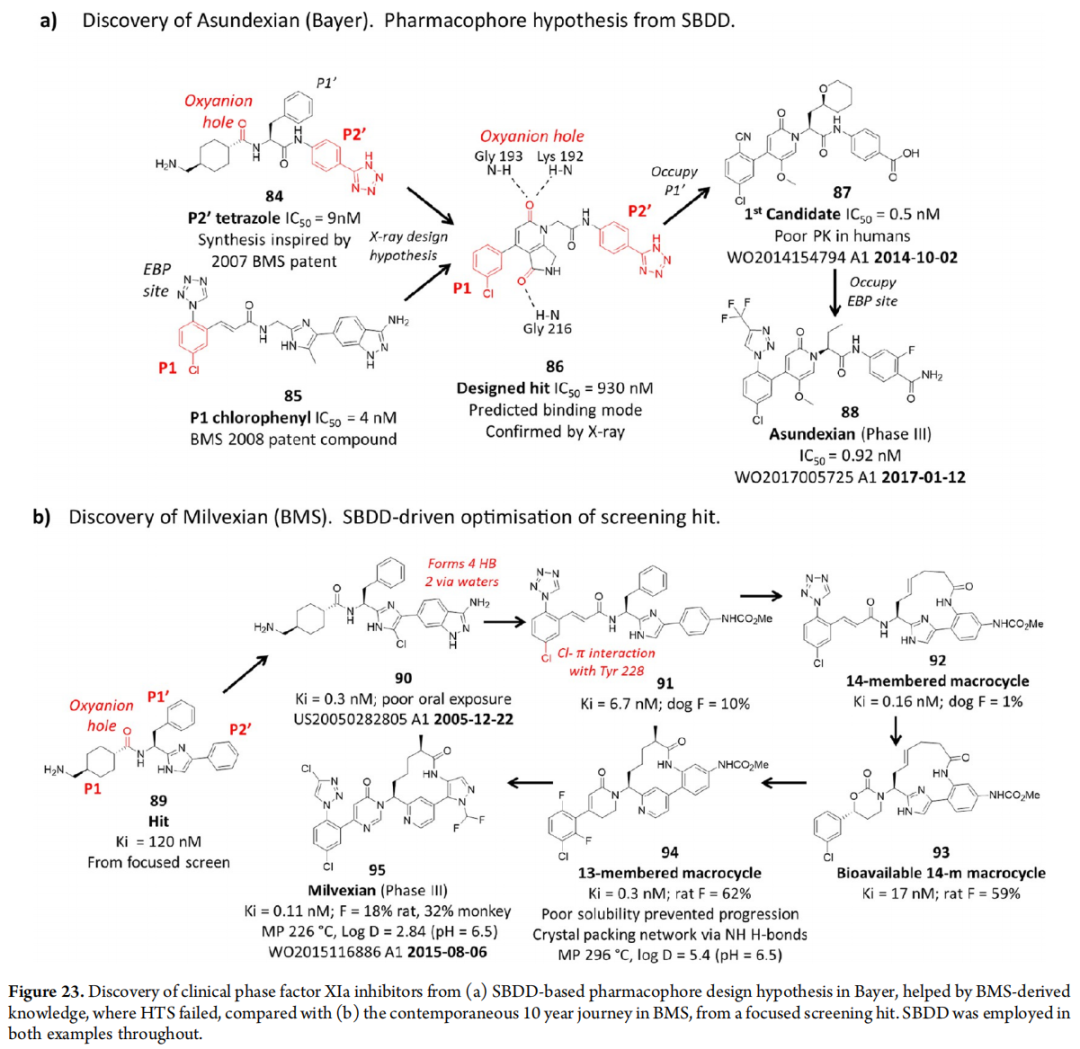
10.4 Factor XIa抑制剂——Asundexian vs Milvexian
两条平行路线的对比极具教育意义:
- • Asundexian(88,Bayer):基于SBDD药效团假设,利用BMS专利知识,在HTS失败背景下设计成功,优化历程相对短暂
- • Milvexian(95,BMS):从聚焦筛选苗头(89)出发,耗时约10年,经过6篇发表论文的系统优化,最终形成大环结构
两个候选药物的"左侧"联芳基三嗪结构高度相似,体现了专利公开带来的知识共享效应。
十一、关键方法论工具的评述
11.1 MCE-18复杂度指标
MCE-18在本研究中是单一最有影响力的优化指标(增加概率85%,归一化变化量最大)。与Fsp³相比,MCE-18额外强调环状脂肪特征和立体化学,更能反映现代药物化学的实践趋势。
11.2 LLE / LipE的核心地位
LLE = p(activity) − XLogP3,在全数据集中平均提升1.6。LLE可理解为靶点结合相对于非特异性膜结合的选择性度量,其系统性提升是优化质量的最佳综合体现。
11.3 bRo5分子的新型表征参数
参数 | 说明 |
|---|---|
EPSA(实验极性表面积) | SFC色谱测量,反映溶剂暴露极性,比TPSA更准确 |
回旋半径(Rg) | 构象系综分析,比MW更适合描述Protac的"尺寸" |
最小最大分子内距离 | 预测Protac膜通透性的新指标 |
EPSA/TPSA比值 | 量化分子变色龙性 |
十二、Lead-like概念的再评价
经典lead-like标准(1999年,Teague等):MW<350,cLogP<3,p(activity)<7。然而本研究数据显示:
- • 仅23%的筛选苗头满足MW<350且cLogP<3(历史数据集中为58%)
- • 40.5%的筛选苗头MW>350且cLogP>3
这一"分子膨胀"现象反映了:新靶标(蛋白-蛋白相互作用、变构位点等)固有的结合口袋特征要求更大、更亲脂的配体,传统lead-like标准已不适用于这些体系。
作者倾向于将lead-like理解为:起始化合物应具备在分子量、复杂度和常见环系向候选药物"靠拢"的潜力,同时维持对cLogP、HBD和芳香环数的控制能力。
十三、对AI/ML的客观评估
数据集中明确由AI/ML主导发现的候选药物案例极少,这与2018—2024年该技术尚处于工具化阶段相符。
作者的核心判断:
"AI/ML工具箱正在快速进化。虚拟筛选可能很快取代HTS,生成化学前景广阔。但候选药物的质量——包括疗效和安全性——才是最终决定药物成败的因素,而这些参数不是AI所能轻易优化的。药物化学家必须主动拥抱并引导AI的发展,而不是被动接受。"
十四、总结与对药物化学实践的启示
核心规律总结
- 1. 分子量增长不可避免,其驱动力是活性提升的需要,应视为正常优化过程
- 2. 亲脂性控制是核心挑战,通过HBA(而非HBD)增加来平衡极性是主流策略
- 3. 脂肪复杂性的增加是当代优化的主旋律,MCE-18是最有价值的单一综合指标
- 4. 苯环向杂芳香环的转换(引入"必要的氮原子")是普遍存在的趋势
- 5. NP片段和PNP骨架的主导地位,提示筛选库设计应强化天然产物来源的三维骨架
- 6. 70%的起始结构被保留,强调起始化合物的选择质量至关重要
- 7. 候选药物的靶标类别特征从起始化合物延续到候选药物,靶标本身是决定性因素
对筛选库设计的建议
基于整体趋势,作者建议未来筛选库应:
- • 低MW,高脂肪复杂性
- • 富含NP片段和PNP骨架
- • 含适量F原子(降低代谢清除风险)
- • 以杂芳香环优先于苯环
- • 在bRo5空间应保持保守,仅在靶标明确需要时进入
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-19,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号