J. Med. Chem. | 面向药物研发的分子表征学习

J. Med. Chem. | 面向药物研发的分子表征学习

DrugIntel
发布于 2026-04-21 11:08:54
发布于 2026-04-21 11:08:54

文献信息
- • 标题:Learning Molecular Representations for Medicinal Chemistry
- • 作者:Kangway V. Chuang, Laura M. Gunsalus, Michael J. Keiser*
- • 期刊:Journal of Medicinal Chemistry, 2020, 63, 8705–8722
- • 机构:加州大学旧金山分校(UCSF)
- • DOI:10.1021/acs.jmedchem.0c00385
一、写在前面:为什么分子表示是核心问题?
在药物发现的计算流程中,有一个常常被忽略、却至关重要的环节——如何将分子"翻译"成计算机可以学习的语言。这个过程,就是分子表示(Molecular Representation)。
无论是经典的 QSAR 建模、虚拟筛选,还是近年火热的 AI 辅助药物设计,所有的预测性能最终都高度依赖于分子表示的质量。一个糟糕的表示,再强大的模型也无从施展;而一个优秀的表示,甚至可以让简单的线性模型取得令人惊讶的效果。
本文是 2020 年发表于 J. Med. Chem. 的一篇 Miniperspective,由 UCSF Keiser 实验室撰写,系统综述了从传统分子描述符到深度学习表示的演进历程,并对多个关键方向进行了深入的批判性分析。时至今日,这篇文章仍是进入"分子机器学习"领域最值得精读的入门综述之一。
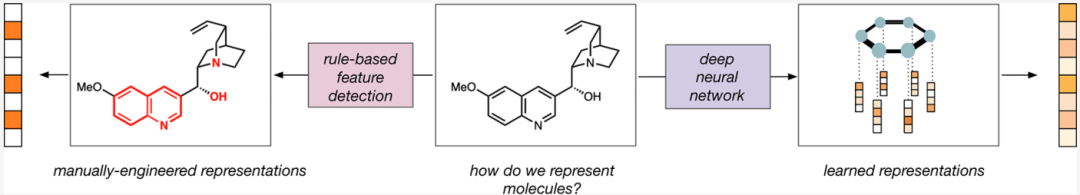
二、什么是分子表示,为什么选择它如此关键?
2.1 表示与学习的关系
作者借助一个经典的机器学习思想实验来说明表示的作用(对应原文 Figure 2):
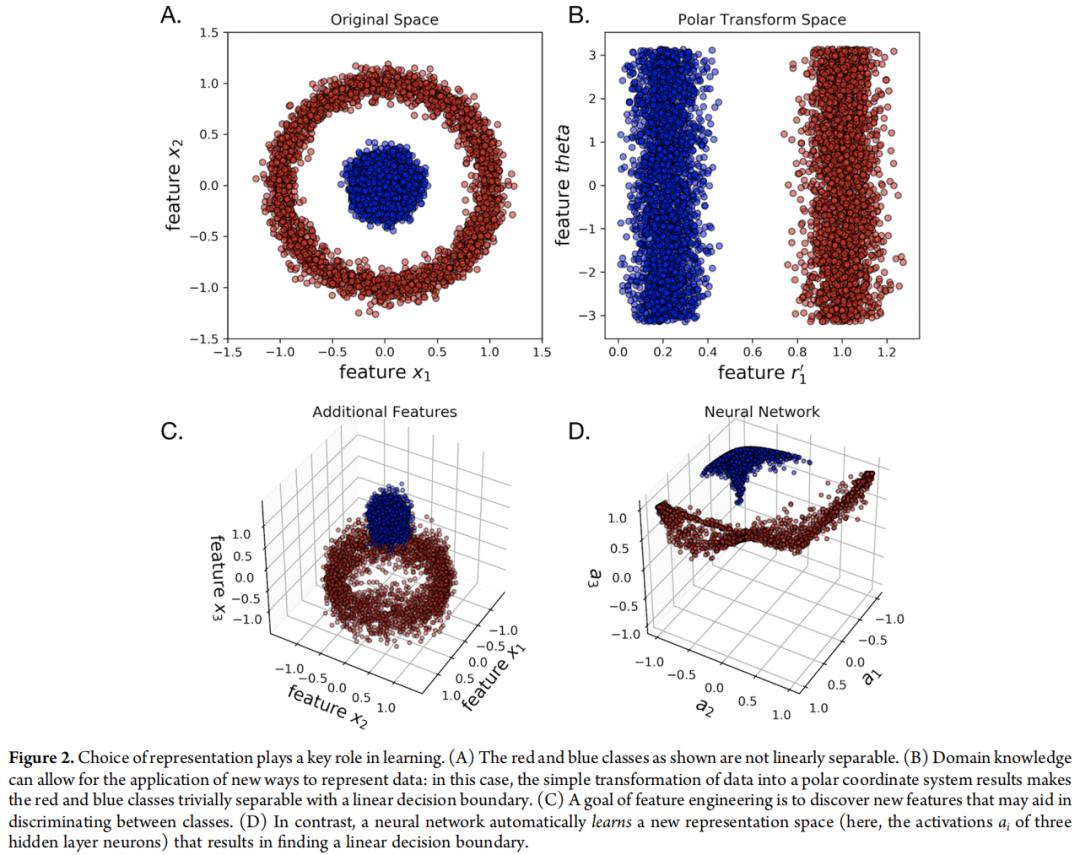
- • 在笛卡尔坐标系下,红蓝两类数据点以同心圆形式分布,线性不可分;
- • 将其转换为极坐标后,两类数据立即变得线性可分;
- • 神经网络的本质,正是自动学习这种坐标变换——在其内部隐层空间中,自动构造出有利于分类或回归任务的新表示。
这一类比精准地揭示了为什么"特征学习(Feature Learning)"比"特征工程(Feature Engineering)"更具潜力:它不依赖人类专家事先定义哪些特征重要,而是让数据自己说话。
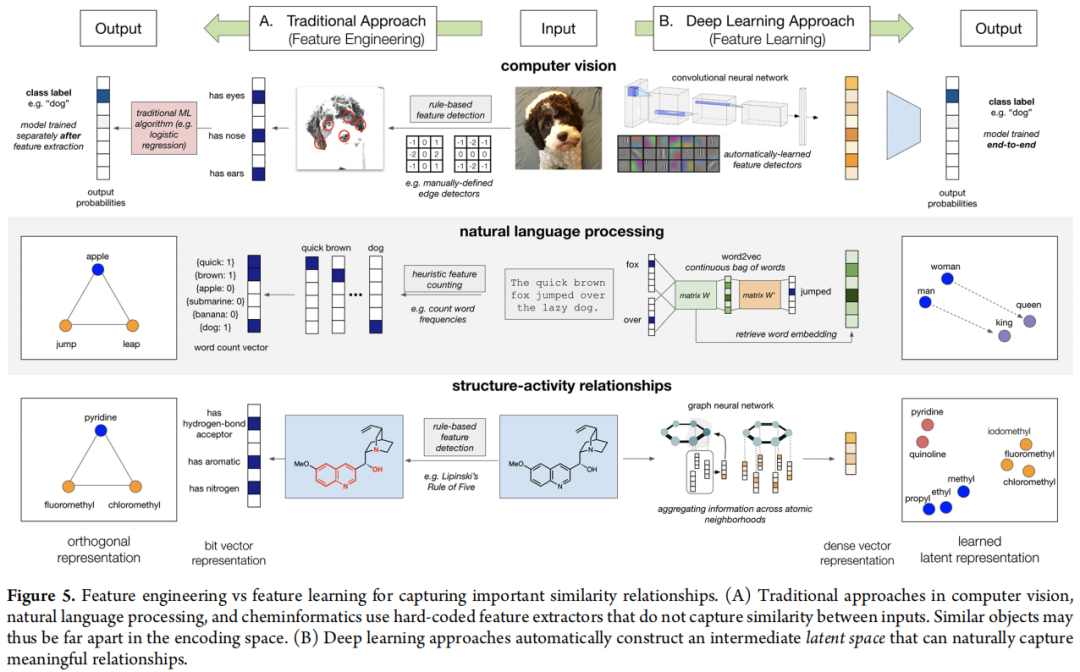
2.2 分子表示面临的独特挑战
分子数据具有以下几个令通用机器学习方法"水土不服"的特性:
挑战 | 具体表现 |
|---|---|
变长输入 | 分子大小不一,难以直接用固定长度向量表示 |
非欧几里得结构 | 分子本质是图(Graph),标准 CNN 无法直接处理 |
组合空间极大 | 类药分子空间估计超过 10^60,远超任何数据集 |
分布外泛化需求 | 药物优化必须探索新结构,模型需对 out-of-distribution 样本有效 |
标注数据稀缺 | 一个典型 QSAR 数据集仅有数百至数千个标注分子 |
最后一点尤为重要:作者明确指出,药物发现从根本上违背了机器学习的 i.i.d. 假设。真正有价值的模型,必须能泛化到训练集之外的化学空间——这对传统机器学习和深度学习都是严峻挑战。
三、分子表示的历史演进
3.1 字符串表示:存储驱动的起点
最早的分子表示方案以信息存储与检索为首要目标:
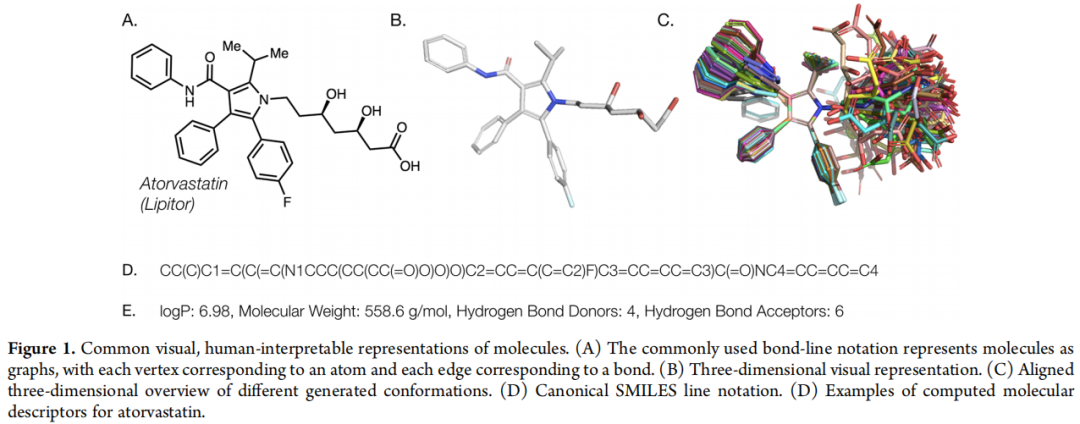
- • SMILES(1988):Simplified Molecular Input Line Entry System,将分子图序列化为字符串,如阿托伐他汀(Lipitor)表示为
CC(C)c1c(...)。紧凑、可读、被广泛采用,是至今最主流的分子线性表示。 - • InChI(IUPAC):国际化学标识符,更强调标准化和唯一性,适合数据库索引。
这些表示是为人类和数据库设计的,并非为机器学习量身定制,后续在深度学习中的应用引入了诸多工程挑战。
3.2 分子指纹:为相似性分析而生
随着 QSAR 研究的兴起,研究者需要将"分子相似性"量化。位串指纹(Bitstring Fingerprints)应运而生,核心思想是:将分子的结构特征编码为固定长度的二进制或整数向量。
几类重要指纹的对比如下:
指纹类型 | 代表方法 | 核心逻辑 | 主要用途 |
|---|---|---|---|
键值型(Key-based) | MACCS Keys(166 位) | 预定义子结构的存在/缺失 | 子结构搜索、药库筛选 |
拓扑型(Topological) | RDKit FP, Atom Pairs | 原子对之间的路径特征 | 相似性搜索 |
圆形(Circular) | ECFP/FCFP | 以每个原子为中心,迭代聚合局部环境 | QSAR、虚拟筛选 |
药效团型 | Pharm2D | 药效团特征的空间分布 | 3D 相似性 |
三维型 | ROCS, Shape-it | 分子形状与静电势 | 骨架跃迁 |
其中,ECFP(Extended Connectivity Fingerprint) 是目前最广泛使用的指纹,由 Rogers & Hahn 于 2010 年系统化描述。其算法与图神经网络的消息传递框架高度对应(详见第五节),是理解 GNN 的重要桥梁。
值得注意的是:尽管 3D 指纹理论上包含更丰富的构象信息,但由于生物活性构象通常未知,且编码单一最低能量构象会引入偏差,3D 方法在实际 QSAR 任务中常被 2D 指纹方法超越——这是该领域的一个重要经验教训。
3.3 描述符:显式物化信息的编码
与指纹并行的另一类表示是分子描述符(Molecular Descriptors),直接编码物理化学性质,如分子量、logP、氢键供/受体数量、拓扑极性表面积(TPSA)等。
描述符的优势在于可解释性强(每一维都有明确物理含义),且计算成本低。Lipinski 五规则即基于少数描述符构建的经验性药代动力学预测框架,历经 30 年仍广泛使用。
四、好的分子表示需要满足哪些条件?
作者提炼出四个核心标准,这是评价任何分子表示方案的理论框架:
4.1 表达性(Expressive)
分子表示必须能捕捉化学空间的丰富多样性,同时能区分细微的结构差异。这里"细微"的含义远超直觉:
- • 质子化状态:同一骨架在不同 pH 下可呈现截然不同的生物活性
- • 互变异构体(Tautomers):酮式与烯醇式可导致完全不同的结合模式
- • 立体化学:对映体可具有完全相反的药理活性(如沙利度胺的 R/S 异构体)
这些"单原子/单键"级别的扰动,在现有分子表示中仍是开放性挑战。
4.2 简洁性(Parsimonious)
奥卡姆剃刀原则在此同样适用。药物化学数据集规模通常有限,高维稀疏的特征空间容易导致维度灾难与过拟合。
ECFP 的长度通常为 1024 或 2048 位,但有效位(非零位)往往只有几十到几百位——冗余极高。深度学习所学的连续向量表示(通常为 128–512 维的密集嵌入),在这一维度上理论上更为高效。
4.3 不变性(Invariant)
分子的图结构对以下变换应保持不变:
- • 原子编号顺序:同一分子的不同编号方式应产生相同表示
- • 旋转与平移(对 3D 方法):分子在空间中的朝向不影响其化学性质
- • 镜像翻转(对手性不敏感任务)
ECFP 通过以每个原子为局部参考系来保证排列不变性;图神经网络则通过置换不变(Permutation-Invariant)的聚合操作(如求和、求均值)来实现这一特性。
4.4 可解释性(Interpretable)
这是在科学应用中最容易被忽视但极其重要的维度。一个纯粹追求预测精度的"黑盒"模型在实际药物发现中风险极高:
- • 模型可能在学习混杂变量(如化合物来源、测试日期、实验批次偏差),而非真实的构效关系
- • 无法验证模型是否学到了"有意义"的化学规律
- • 不能为药物化学家提供结构优化的方向性建议
五、深度学习的核心架构与分子学习
5.1 为何现在是深度学习的时代?
深度神经网络(DNN)并非新生事物——上世纪 90 年代就已应用于 QSAR。然而,它在过去十年的爆发式成功,依赖三个关键要素的协同成熟:
- 1. 数据规模:ChEMBL、ZINC15 等开放数据库积累了数以亿计的分子及生物活性数据
- 2. 算法突破:BatchNorm、ReLU 激活函数、Adam 优化器、残差连接(ResNet)解决了深层网络的训练困难
- 3. 硬件加速:GPU 使并行矩阵运算速度提升 100–1000 倍,大幅降低训练成本
对于分子来说,这三者的结合意味着:我们第一次有能力在足够大的化学数据上训练足够深的网络。
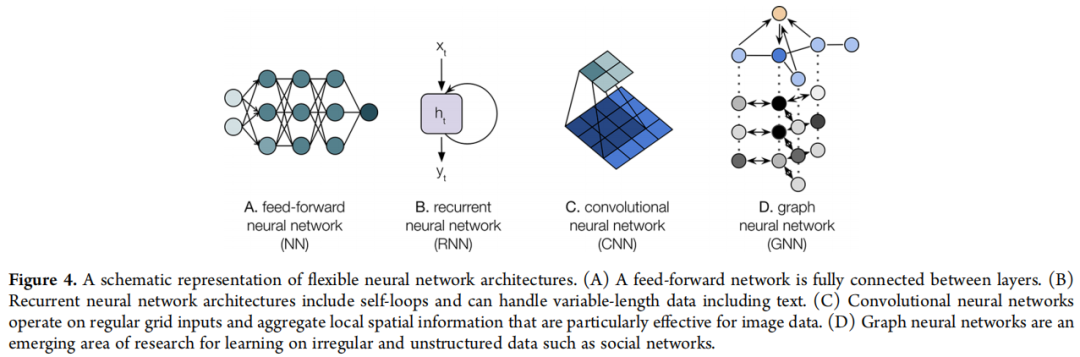
5.2 各类神经网络架构与分子学习的对应关系
架构 | 适用输入 | 核心机制 | 分子应用 |
|---|---|---|---|
全连接网络(FCNN) | 固定长度向量 | 逐层非线性变换 | 经典 QSAR(输入为指纹/描述符) |
循环神经网络(RNN/LSTM) | 变长序列 | 时序隐状态传递 | 从 SMILES 字符串学习分子表示、分子生成 |
卷积神经网络(CNN) | 规则网格 | 局部感受野 + 参数共享 | 分子图像表示(较少用)、1D 序列处理 |
图神经网络(GNN) | 图结构 | 消息传递(Message Passing) | 从原子图直接学习,目前最主流方向 |
Transformer | 序列/集合 | 自注意力机制 | 分子预训练模型(如 ChemBERTa,超出本文范围) |
5.3 图神经网络:从 ECFP 到可学习的"神经指纹"
这是本文最具技术深度的部分,值得细读。
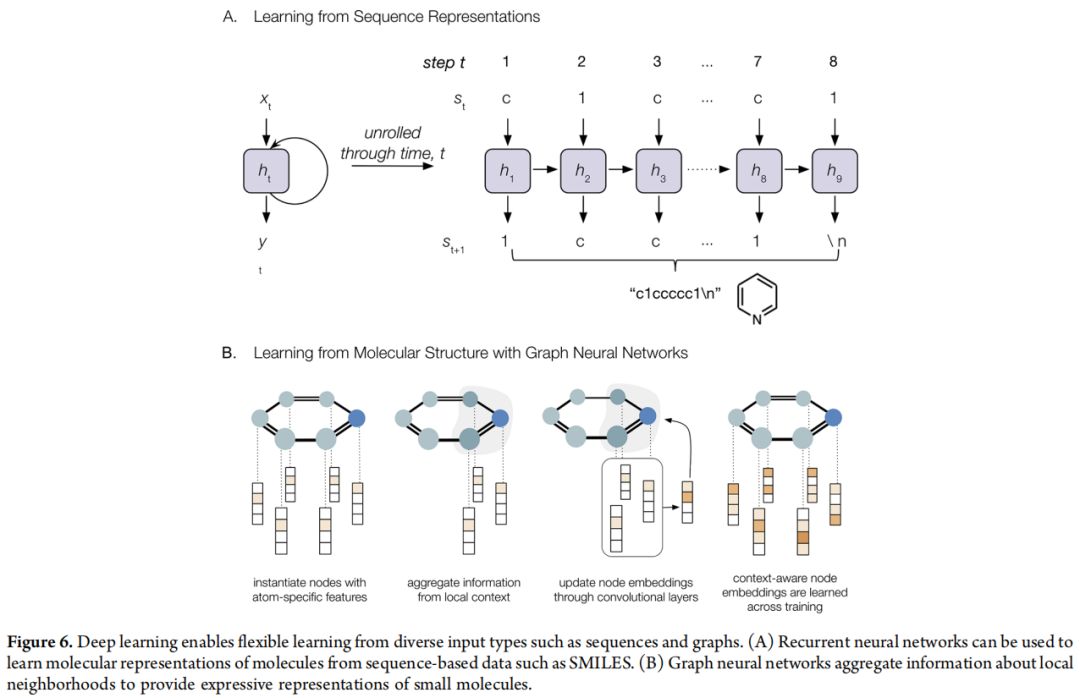
ECFP 的算法流程:
- 1. 为每个原子赋予初始标识符(基于原子类型、度、氢数等)
- 2. 迭代:将当前标识符与邻居标识符通过哈希函数聚合,生成新标识符(对应"半径"r 的圆形邻域)
- 3. 将所有标识符的集合映射到固定长度位串(通过哈希)
GNN 的消息传递框架:
- 1. 为每个原子赋予初始特征向量(原子类型、杂化态、形式电荷等)
- 2. 迭代 次:每个原子从其邻居聚合信息,通过可学习的神经网络层更新自身表示
- 3. 对所有原子表示进行全局聚合(readout),得到分子级向量
两者的结构上完全同构——GNN 本质上就是 ECFP 的可微分、可学习版本。关键区别在于:
- • ECFP 使用固定哈希函数聚合,对任务不可知;GNN 的聚合函数由数据驱动优化,对任务自适应
- • ECFP 产生离散的、稀疏的位串;GNN 产生连续的、密集的嵌入向量,保留了片段间的相似性信息
- • ECFP 中"氯乙基"与"氟甲基"是完全不同的编码;GNN 可学习到它们在功能上的相似性
早期工作中,Duvenaud 等人的神经图指纹(Neural Graph Fingerprints) 和 Kearnes 等人的分子图卷积(Molecular Graph Convolutions) 证明了 GNN 可以在水溶性和生物活性预测任务上达到或超越 ECFP 的性能,开创了这一方向的先河。
六、深度学习的四大机遇:技术深度解析
6.1 从灵活输入格式学习
SMILES 的局限性是一个重要的工程问题,文章给出了三点批评:
- 1. 非唯一性:同一分子可以有多个合法 SMILES 字符串(如苯可以写成
c1ccccc1也可以写成c1cccc c1),导致训练数据中存在同义不同形的噪声 - 2. 脆弱性:单个字符的改动可能产生化学上无效的分子
- 3. 线性化失真:分子本质是非线性的图,强行序列化必然损失拓扑信息
针对这些问题,研究者提出了多种解决方案:
- • SMILES 数据增强(Bjerrum, 2017):对同一分子使用多种合法 SMILES 表示作为训练样本,提高模型鲁棒性
- • DeepSMILES(O'Boyle & Dalke, 2018):修改 SMILES 语法,减少不合法字符串的产生
- • SELFIES(Krenn et al., 2019):自参照嵌入字符串,数学上保证任意随机字符串都对应一个合法分子,根本解决生成有效性问题
- • 图神经网络:直接在图结构上操作,绕开字符串表示的所有问题
6.2 连续化学空间与分子相似性的重新定义
相似属性原则(Similar Property Principle) 是 QSAR 的基石:结构相似的分子应具有相似的性质。然而,Tanimoto 系数等传统相似性度量有根本缺陷:
- • 活性悬崖(Activity Cliffs):两个 Tanimoto 系数高达 0.9 的分子,活性可能相差 1000 倍以上
- • 骨架跃迁(Scaffold Hops):两个结构截然不同(Tc < 0.3)的分子可以作用于同一靶点,且具有相似效力
这两种情况在传统指纹空间中都是"异常值",但在功能上分别代表"过高估计相似性"和"过低估计相似性"。
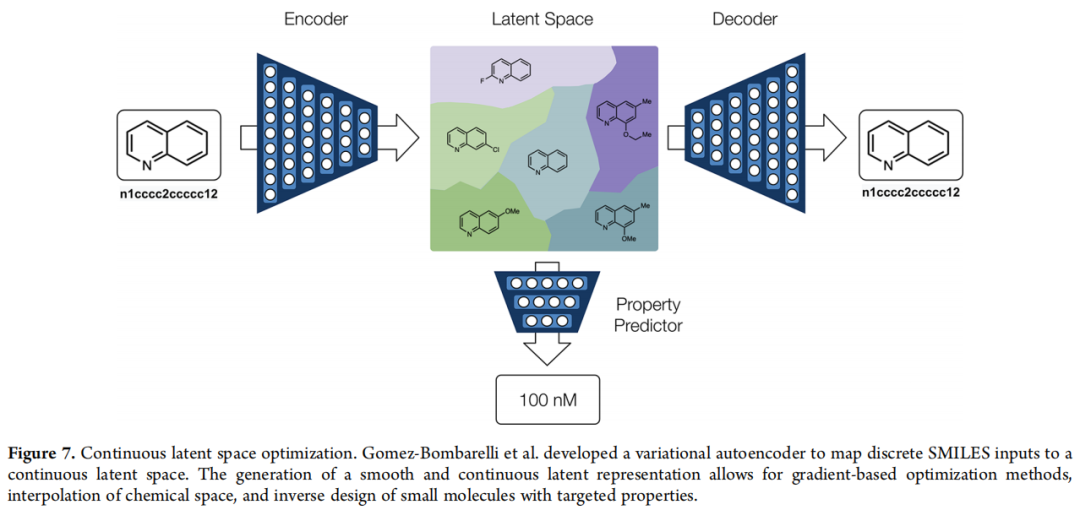
深度学习的连续嵌入空间从理论上提供了更优越的解决方案:网络可以在训练过程中,将功能相似的分子映射到嵌入空间的邻近区域,而与其拓扑相似性无关。Gomez-Bombarelli 等人的 VAE 工作 是这一思想的最佳实践:
- • 编码器:将 SMILES 字符串压缩为低维连续潜向量(256 维)
- • 解码器:从潜向量重建 SMILES 字符串
- • 性质预测网络:与编码器联合训练,使潜空间具有"性质可预测"的结构
- • 梯度优化:直接在连续潜空间中沿着性质梯度方向移动,实现分子优化
这使得"分子优化"从组合问题变为连续优化问题,可以使用成熟的梯度下降算法。
6.3 生成模型与从头药物设计
逆向 QSAR(Inverse QSAR/QSPR) 是药物设计的圣杯:给定目标活性,生成具有该活性的分子结构。这一问题的难点在于:
- • 分子空间是离散的,无法直接应用基于梯度的优化
- • 活性预测模型(正向)的输出空间(实数)与分子生成的输出空间(图/字符串)存在本质鸿沟
深度生成模型提供了三种主流框架:
① 变分自编码器(VAE)
- • 核心贡献:将离散分子空间连续化
- • 代表工作:Gomez-Bombarelli et al.(ACS Cent. Sci. 2018);Jin et al. 的 Junction Tree VAE(ICML 2018)
- • 后者通过在分子片段树(Junction Tree)层面操作,从根本上保证了生成分子的化学有效性
② 循环神经网络(RNN)生成模型
- • 类比语言模型,逐字符生成 SMILES
- • 代表工作:Segler et al.(ACS Cent. Sci. 2018)的两步迁移学习方法:先在 140 万个 ChEMBL 分子上预训练,再在已知活性分子上微调,生成聚焦化合物库
- • Popova et al. 使用 stack-RNN 实现 95% 的结构有效率
③ 图生成网络
- • 直接生成分子图,绕开字符串表示的问题
- • 代表工作:MolGAN(De Cao & Kipf, 2018)使用生成对抗网络(GAN)
- • 图的生成比编码更具挑战性(节点/边需要逐步决策),但图有效率更高
作者的冷静批判值得关注:
Zhavoronkov 等人(Nat. Biotechnol. 2019)用深度生成模型发现 DDR1 激酶抑制剂,最优化合物 IC₅₀ 达 10 nM,整个流程仅用数周——听起来令人振奋。然而 Walters & Murcko 的评论指出,该最优化合物与已知 DDR1 抑制剂高度相似,新颖性存疑。这一争论点出了生成模型的核心困境:如何在新颖性(Novelty)与可合成性(Synthesizability)之间取得平衡?
此外,Stokes 等人(Cell 2020)用虚拟筛选(而非生成模型)发现了广谱抗生素 Halicin,同样令人印象深刻——说明枚举式筛选与生成式设计在实践中仍是互补而非替代的关系。
6.4 多任务学习与迁移学习
多任务学习(Multitask Learning) 的核心假设:多个相关任务共享底层的化学规律,联合训练可让模型在学习任务 A 的同时,借助任务 B 的信号来改善任务 A 的表示学习。
从信息论角度理解:对于单任务模型,只有正比于数据集大小 N 的"有效样本量";多任务模型通过辅助任务,相当于为每个原始任务引入额外监督信号,等效扩大了训练集。
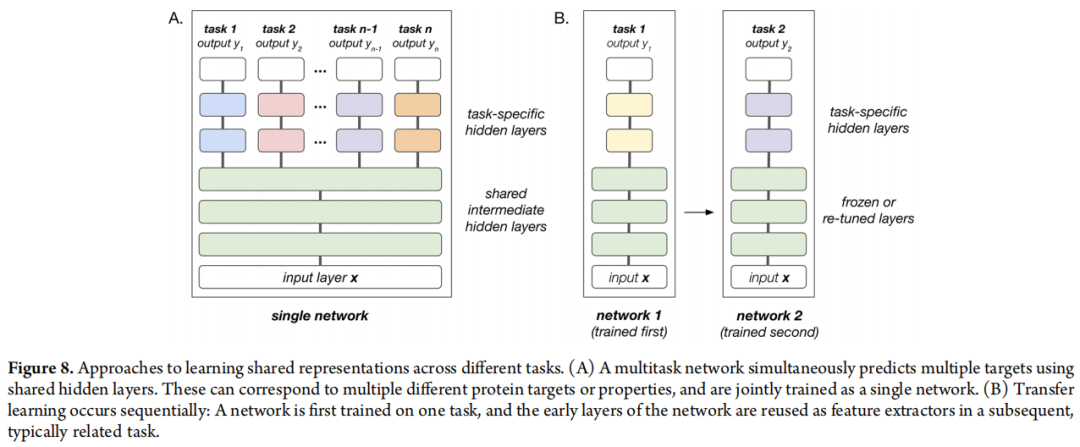
实证证据的梳理:
研究 | 发现 |
|---|---|
Dahl et al. (Merck Kaggle 冠军) | 多任务 DNN 比单任务随机森林平均提升约 15% |
Ma et al. (Merck 内部验证) | 相对随机森林改善更为有限,约 1–2% |
Ramsundar et al. | 跨数百个蛋白靶点多任务学习,总体有小幅提升 |
Xu et al. | 明确记录了负迁移(Negative Transfer)现象:添加不相关辅助任务会损害目标任务性能 |
Rodríguez-Pérez & Bajorath | 103 个激酶靶点多任务一致改善——任务相关性是关键 |
结论:多任务学习并非万能,任务相关性是成败的决定因素。激酶家族内多任务天然适配(共享 ATP 结合口袋特征),而跨蛋白家族的多任务则需要谨慎评估。
迁移学习(Transfer Learning) 在计算机视觉中的成功有目共睹:ImageNet 预训练的 ResNet 被迁移到皮肤癌分类,仅用数百张图片即可达到皮肤科专家水平(Esteva et al., Nature 2017)。
然而,在分子学习中直接复制这一成功面临更大困难:
- • 化学空间的多样性远超图像空间:猫和狗共享大量视觉特征(边缘、纹理),而激酶与GPCR之间的底层化学规律差异极大
- • 数据规模不匹配:ImageNet 有 120 万张图,而 ChEMBL 高质量活性数据仅约 50–100 万条,且分散在数千个靶点上
- • 负迁移风险:Hu et al.(arXiv 2019)发现,GNN 迁移学习中,预训练策略的选择(节点级 vs. 图级、有监督 vs. 无监督)对迁移效果影响极大,不当的预训练甚至导致性能下降
七、模型可解释性:药物发现中不可或缺的维度
7.1 为何可解释性在科学应用中尤为重要
作者引用了 McCloskey et al. 一个极具说服力的反例:在人工构造的、真实活性已知的合成数据集上,神经网络通过学习数据集中的混杂相关性(而非真实的构效关系),仍能在测试集上取得近乎完美的准确率。
这表明:高 AUC-ROC 或高 R² 并不等于模型学到了有意义的化学知识。若不进行可解释性分析,强大的模型可能只是在"记住"数据集的偏差。
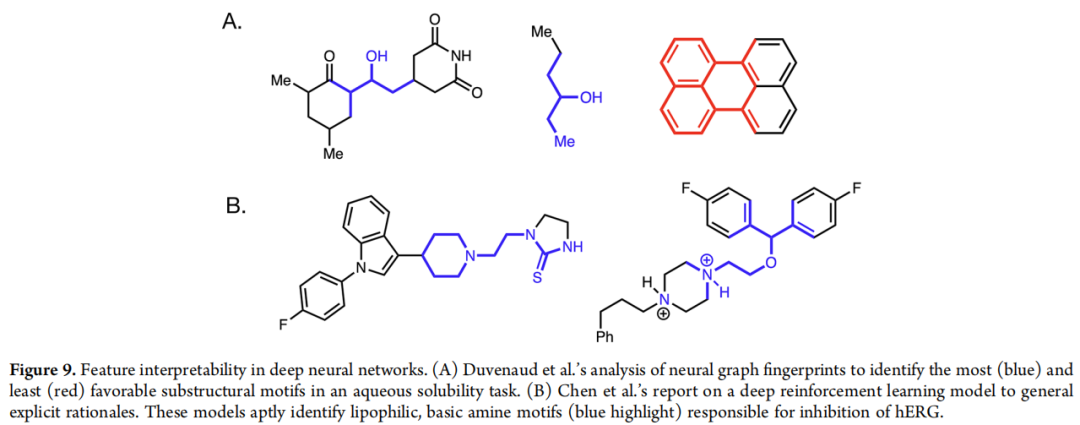
7.2 可解释性方法工具箱
事后归因方法(Post-hoc Attribution):
- • 特征消融(Feature Ablation):系统性地移除某个特征,观察预测变化;简单但可能给出误导性结果(Sheridan, 2019)
- • 梯度显著图(Gradient Saliency Maps):计算输出对每个输入特征的梯度,高梯度区域对预测贡献大;对应 GNN 中的原子重要性热图
- • 积分梯度(Integrated Gradients, IG):沿从基准点到输入点的路径积分梯度,理论上满足完备性公理,更可靠
- • 注意力权重(Attention Weights):在 Transformer 类模型中,注意力分数可视化哪些原子/片段对预测影响最大
本征可解释模型(Intrinsically Interpretable):
- • Chen et al. 的深度强化学习方法:训练模型同时生成预测和预测理由(显式子结构高亮),对 hERG 抑制预测中,模型准确识别出亲脂性碱性胺基团——与电生理实验证据高度吻合
7.3 作者的审慎态度
文章明确指出现有解释性研究的局限:
- • 多数案例是确认性的——验证模型学到了已知化学规律(如羟基→水溶性,碱性胺→hERG 阻断),而非发现新的 SAR
- • 容易陷入确认偏误:研究者可能选择性展示与直觉一致的例子
- • 使用不同可解释性方法对同一模型分析,结果可能相互矛盾
因此,作者倡导假设驱动的可解释性研究:先提出具体的、可证伪的化学假设,再用可解释性工具验证模型是否学到了对应的模式,而非在训练完成后进行无结构的"探索"。
八、深度学习的现实局限:清醒的批判视角
局限性 | 详细分析 |
|---|---|
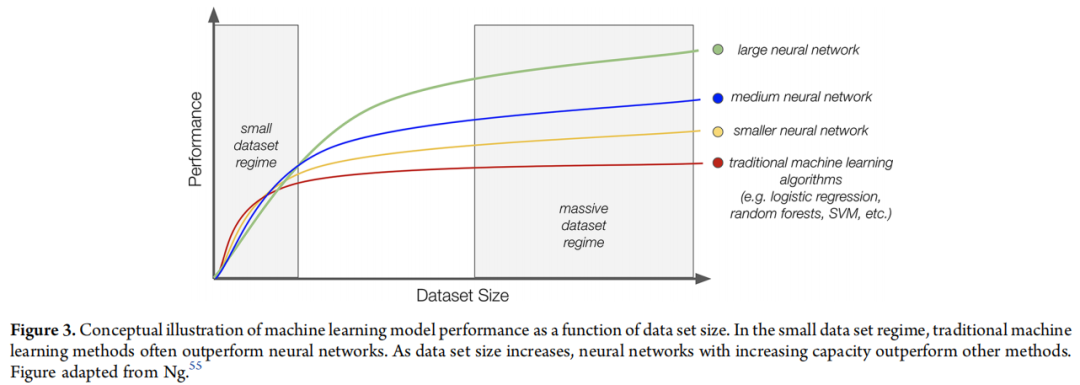
数据瓶颈 | DNN 在 massive data regime 才能充分发挥优势(Figure 3)。许多 QSAR 任务数据集仅数百至数千条,传统 ECFP + RF/SVM 仍是基准线难以超越的实用选择 |
数据质量 | ChEMBL 等聚合数据库存在出版偏差(阳性结果过多)、测量方法异质性、数量级跨度大等问题,高噪声数据损害 DNN 训练稳定性 |
训练成本 | ECFP 计算一个分子不到 1 毫秒;训练一个 GNN 模型可能需要数小时至数天,且超参调优成本高 |
使用门槛 | 需要 Python/PyTorch/DGL 等技能;模型的正确评估需要专业知识(正确划分数据集、避免数据泄露等) |
可重复性 | 随机初始化、数据集随机划分、不同框架实现的微小差异都可能显著影响结果;许多论文未开源代码或数据 |
分布外泛化 | 当前 DNN 在化学空间"内插"表现良好,但在"外推"——即与训练分子结构显著不同的新骨架——上可靠性仍存疑 |
九、未来方向展望
作者在结论部分指出了几个最值得关注的前沿方向:
① 三维结构与构象动态的有效编码
现有方法主要处理 2D 拓扑(静态分子图)。然而,分子识别(如蛋白-配体结合)本质上是三维事件,且涉及柔性分子的构象集成(Conformational Ensemble)。如何有效编码三维信息而不引入构象偏差,仍是开放问题。等变图神经网络(Equivariant GNN,如 SE(3)-Transformers、SchNet、DimeNet)是目前最有前景的方向之一(但超出本文范围)。
② 整合结构生物学的多模态学习
将小分子表示与蛋白质序列/结构表示(如 AlphaFold 预测的结构)在同一框架下联合学习,实现真正的配体-靶点共建模,是下一代虚拟筛选的重要方向。
③ 主动学习与实验闭环
深度学习模型与实验测试之间的闭环(Active Learning / Bayesian Optimization),可以用最少的实验资源最快速地缩小候选分子空间——这比大规模虚拟筛选或生成式设计在实践中可能更为高效。
十、综合评价与阅读建议
本文的核心贡献
- 1. 系统梳理了从传统指纹到深度学习的分子表示演进,建立了清晰的概念框架
- 2. 深度解析了 GNN 与 ECFP 的算法对应关系,帮助读者从已知出发理解 GNN
- 3. 批判性评估了生成模型、多任务学习、迁移学习的现实局限——这种清醒在该领域并不常见
- 4. 强调可解释性的科学重要性,并给出具体的方法论建议
本文的时代背景与延伸阅读
本文成文于 2020 年,Transformer/BERT 类预训练语言模型尚未在分子领域全面铺开。此后,ChemBERTa、MolBERT、Uni-Mol 等大规模分子预训练模型相继出现,在多项基准任务上大幅刷新了 GNN 的性能记录——这是本文未能覆盖但同样重要的进展。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-16,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号