AlphaFold 不只是预测结构:它如何把蛋白 binder 设计成功率提高近 10 倍?

AlphaFold 不只是预测结构:它如何把蛋白 binder 设计成功率提高近 10 倍?

DrugIntel
发布于 2026-07-15 14:28:10
发布于 2026-07-15 14:28:10

文献来源
论文题目: Improving de novo protein binder design with deep learning 期刊: Nature Communications 发表时间: 2023 年 通讯作者: David Baker 论文类型: 方法改进论文 + 回顾性分析 + 前瞻性实验验证论文 核心贡献: 作者将 AlphaFold2、RoseTTAFold 和 ProteinMPNN 引入传统 Rosetta 蛋白 binder 设计流程,用结构预测模型识别不会按预期折叠或不会按预期结合的设计,并用 ProteinMPNN 提升序列设计效率,最终在新靶点前瞻性实验中显著提高设计成功率。
导读
de novo 蛋白 binder 设计的目标,是仅根据靶蛋白结构,设计出能够特异性结合目标表面区域的新蛋白。传统 Rosetta 方法已经证明这一路线可行,但实验命中率很低,需要筛选大量候选序列。Baker 团队这篇论文的关键思路不是重新发明整个生成模型,而是把设计失败拆成两个具体问题:序列是否能折叠成设计结构,设计结构是否能形成预期复合物。作者用 AlphaFold2 和 RoseTTAFold 评估这两个环节,并发现复合物界面 pAE_interaction 是非常有效的成功率过滤指标;进一步结合 ProteinMPNN 替代 Rosetta 进行序列设计,使流程更高效。文章的价值在于,它把结构预测模型从被动验证工具变成了蛋白设计流程中的主动质量控制模块。
为什么这篇论文值得关注?
蛋白 binder 是一类能够与目标蛋白特定表面结合的蛋白质分子,可以用于治疗、诊断、成像、靶向递送和基础生物学研究。传统获得 binder 的方式主要依赖免疫动物、抗体库筛选、酵母展示或噬菌体展示。这些方法成熟可靠,但往往需要较大实验投入,而且对结合位点、分子大小、稳定性、表达性和可工程化性质的控制有限。
de novo 计算设计的理想目标,是只给定靶蛋白结构和希望结合的表面区域,计算机就能生成一个全新的小型蛋白,使其能够稳定折叠,并以预期构象结合目标位点。这个目标非常有吸引力,因为它理论上可以让研究者直接指定结合表面,快速获得抗体之外的新型亲和试剂。
但真正困难的地方在于:设计出来的蛋白不只是要看起来能结合,还必须在真实物理环境中完成两个动作。第一,它自身要折叠成设计者希望的结构;第二,它要以设计者希望的方式和靶蛋白形成复合物。任一环节失败,候选 binder 都会在实验中变成阴性。
这篇论文真正值得关注的地方,正是它没有把失败笼统归因于模型不够强,而是把失败拆成可诊断、可过滤、可优化的两个层级,并用深度学习结构预测模型分别处理。
研究背景
传统 Rosetta 蛋白 binder 设计的基本逻辑,是在靶蛋白表面指定一个目标区域,然后通过骨架放置、界面设计、序列优化和能量过滤,寻找同时满足两个条件的候选分子:
一方面,binder 序列在游离状态下应当以设计骨架为最低能量构象;另一方面,binder 与靶蛋白形成复合物时,界面能量应当足够有利,能够驱动结合。
Rosetta 的优势在于物理可解释性强,能够显式考虑范德华相互作用、氢键、电荷、溶剂化和键合几何等因素。问题是,为了计算效率,Rosetta 能量函数通常被写成若干可分解项的加和,这种近似很难完整描述蛋白折叠与蛋白-蛋白界面的真实自由能景观。与此同时,binder 设计需要搜索的构象、序列和界面空间极其庞大。如果能量函数不够准确,或者采样不够充分,就可能把错误构象评估为好设计。论文在第 2 页将这一点说得很清楚:低成功率主要来自能量函数误差与巨大搜索空间共同造成的折叠失败和结合失败。
作者把失败分成两类:
Type I failure:折叠失败。 设计序列不能折叠成设计时假定的 binder 单体结构。也就是说,计算模型认为这个序列对应某个小型螺旋束或折叠骨架,但真实情况下它可能折叠到另一个状态,或者根本不稳定。
Type II failure:结合失败。 设计序列可以折叠成预期单体结构,但这个单体并不会按照设计模型与靶蛋白结合。也就是说,单体是对的,界面是错的。
论文 Figure 1 用非常直观的方式展示了这两个失败模式:成功 binder 必须先从序列折叠到设计单体,再从设计单体形成设计复合物;Type I 卡在折叠,Type II 卡在界面。这个拆分是全文的关键逻辑。
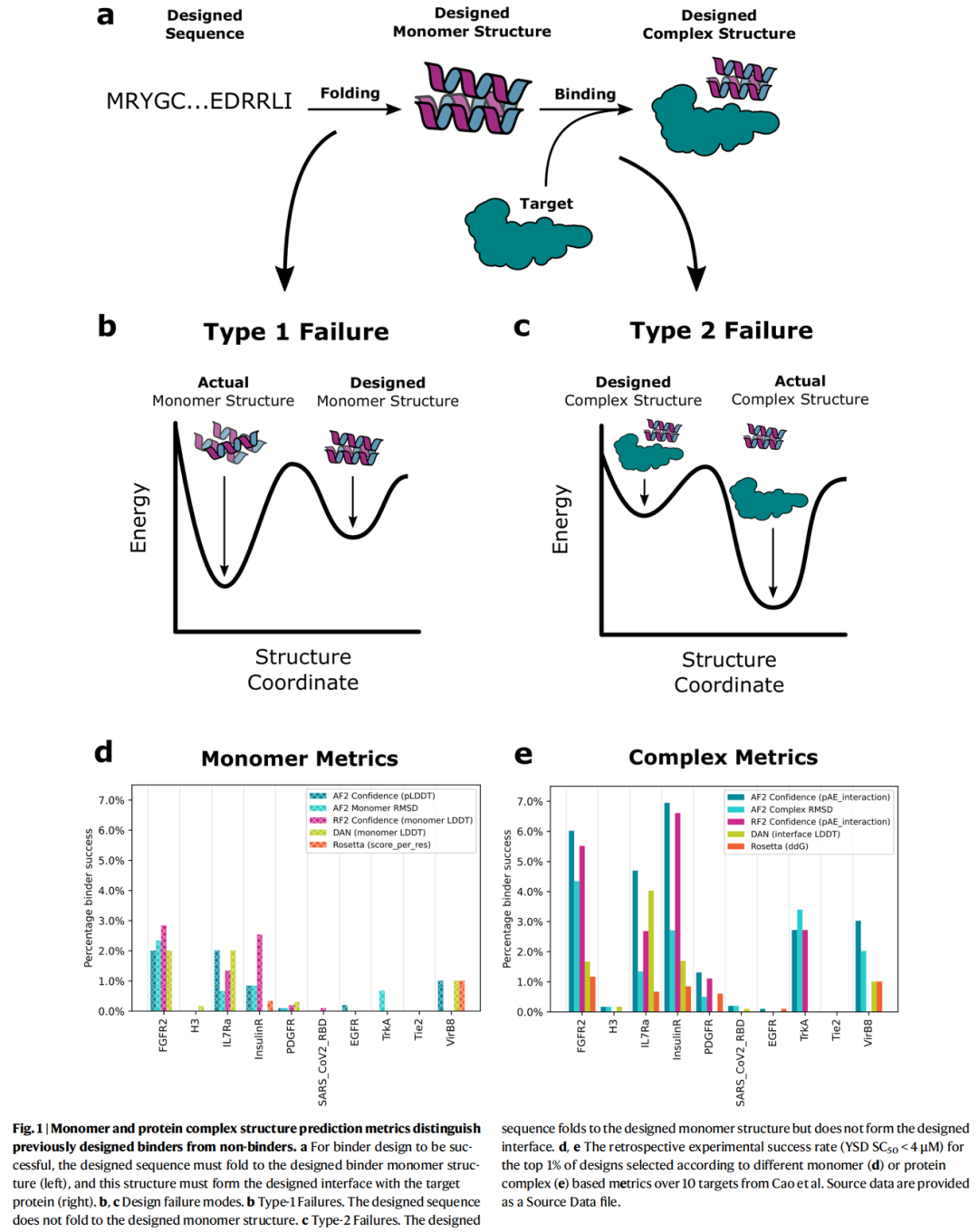
3. 以往方法
3.1 实验筛选方法:可靠,但不够可控
抗体免疫、抗体库筛选和其他 scaffold library 筛选能够产生高亲和 binder,但实验成本高,而且得到什么样的 binder 很大程度上取决于库的构成和筛选压力。对于想要精确靶向某个结构表面、避开某些区域、控制分子大小或提升热稳定性的任务,这类方法的可控性有限。
3.2 Rosetta 设计方法:可控,但命中率低
Rosetta-based binder design 的优势在于可以从靶点结构出发,围绕指定表面区域设计全新蛋白。它提供了一条从结构到序列再到实验验证的完整路线。
但问题是,Rosetta 主要依赖物理能量函数和采样流程来判断设计是否合理。对于单体折叠,模型要判断序列是否对应设计结构;对于界面结合,模型要判断复合物是否足够稳定。两者都涉及复杂能量景观,仅靠局部能量优化和有限采样很难完全可靠。
3.3 深度学习结构预测:强在全局结构一致性判断
AlphaFold2 和 RoseTTAFold 的能力不只是预测天然蛋白结构。对于 de novo 设计蛋白,由于序列通常更规则、更稳定、更接近工程化折叠单元,单序列输入也常常能得到较准确的结构预测。
这给 binder 设计提供了一个新机会:不再只问 Rosetta 能量是否低,而是让结构预测网络判断这个序列是否真的倾向于形成设计结构,以及是否倾向于与靶蛋白形成设计复合物。
这篇文章正是把 AF2/RF 从结构预测器转化为设计过滤器。
这篇论文的核心思想
这篇论文的核心思想可以概括为一句话:
用深度学习结构预测模型检查 Rosetta 设计结果是否自洽,再用 ProteinMPNN 降低序列设计成本。
具体来说,作者做了两件事。
第一,用 AF2/RF2 评估设计序列是否能折叠成设计单体结构。这里主要看 AF2/RF2 预测出的 binder 单体与 Rosetta 设计单体之间的 Cα RMSD 是否小,以及预测置信度 pLDDT 是否高。若预测结构明显偏离设计结构,则说明该序列可能发生 Type I failure。
第二,用 AF2/RF2 评估设计 binder 是否能和靶蛋白形成预期复合物。这里关键指标是复合物预测中的 pAE_interaction,即跨链残基对的平均预测对齐误差。pAE_interaction 越低,表示模型越确信两条链之间的相对位置关系,也就是越确信该 binder 与靶蛋白形成了稳定而明确的界面。
相比单纯使用 Rosetta ddG,pAE_interaction 的意义在于:它不是只评价设计复合物这个局部结构的能量深度,而是让结构预测网络在更大的折叠与结合空间中判断这个序列-结构-界面组合是否可信。
5. 方法细节:模型到底是怎么做的?
5.1 任务定义与输入输出
本文处理的是 one-sided protein interface design,也就是靶蛋白结构给定,设计一个新的 binder 去结合它。所谓 one-sided,是因为靶蛋白一侧通常固定,主要设计 binder 一侧的骨架、序列和界面。
输入包括:
- • 靶蛋白三维结构;
- • 希望结合的靶蛋白表面区域;
- • 可用于 binder 设计的 scaffold 或骨架集合;
- • Rosetta 生成的大规模候选设计;
- • 对于 AF2/RF2 过滤,还需要 binder 序列、靶蛋白结构、设计复合物结构或初始猜测结构。
输出包括:
- • 通过计算过滤的候选 binder 序列;
- • 对应的设计单体结构与设计复合物结构;
- • AF2/RF2 预测结构与置信度;
- • 最终进入酵母展示和 BLI 验证的候选集合。
从实际流程看,这不是一个端到端生成模型,而是一个多阶段设计-预测-过滤-实验验证系统。
5.2 数据表示:序列、单体结构与复合物结构三层表示
这篇论文涉及三类核心表示。
第一是binder 序列表示。对于 AF2 和 RF2,binder 主要以氨基酸序列形式输入。对于 de novo 设计序列,作者使用单序列预测即可评估单体折叠趋势。
第二是单体结构表示。Rosetta 设计会给出 binder 的目标单体结构。AF2/RF2 预测后,作者比较预测单体与设计单体之间的 Cα RMSD,并结合 pLDDT 判断单体结构是否可信。
第三是复合物结构表示。binder 与 target 形成复合物后,作者关注跨链相对几何关系。这里不仅看 AF2/RF2 预测复合物是否接近 Rosetta 设计模型,还重点看 pAE_interaction,即 target-chain 与 binder-chain 残基对之间的预测对齐误差。
pLDDT 主要反映局部结构置信度,适合判断单体折叠是否稳健;pAE_interaction 更适合判断链间相对放置是否可信,因此在界面设计中更关键。
5.3 深度学习过滤模块:AF2/RF2 如何判断设计是否靠谱?
作者首先做回顾性分析,数据来自此前 Cao 等人的大规模实验设计集合:约 100 万个已经实验表征的设计,覆盖 10 个不同靶点。每个靶点实验测试约 1.5 万到 10 万个设计,真实 binder 数量从 1 个到 584 个不等。作者把这些实验结果作为已知标签,比较不同计算指标区分 binder 与 non-binder 的能力。
对于 Type I failure,作者比较了:
- • Rosetta 单体能量,按链长归一化;
- • DeepAccuracyNet 的单体结构准确性预测;
- • AF2/RF2 预测单体与设计单体之间的 Cα RMSD;
- • AF2/RF2 的 pLDDT。
结果显示,Rosetta 单体能量区分能力有限,因为它已经在早期筛选中被用过,再次使用新增信息较少;DAN 有一定区分能力;AF2/RF2 预测结构越接近设计结构,实验成功概率越高;pLDDT 也具有预测作用。
对于 Type II failure,作者比较了:
- • Rosetta ddG;
- • DAN 复合物准确性指标;
- • AF2/RF2 预测复合物与设计复合物之间的 Cα RMSD;
- • AF2/RF2 的 pAE_interaction。
结果显示,pAE_interaction 是最有用的复合物级别过滤指标之一。尤其在 IL7Rα、TrkA、FGFR2、InsulinR 和 PDGFR 等数据集中,低 pAE_interaction 能明显富集实验确认的 binder。作者发现 pAE_interaction < 10 附近会出现成功率的明显上升,因此在后续前瞻性设计中将其作为重要过滤阈值。
5.4 AF2 initial guess:为什么需要给 AlphaFold 一个初始复合物?
直接用 AF2 预测 binder-target 复合物并不总是稳定。作者在五个已有实验结构的 minibinder 上测试时发现,AF2 能正确预测部分复合物,但对于 SARS-CoV-2 Spike 相关设计,AF2 由于没有正确建模 Spike 的一段长 loop,导致 binder 被预测为未结合状态。
为了解决这个问题,作者尝试给 AF2 提供靶蛋白模板,但单独使用 target template 会带来新的问题:目标蛋白结构对了,界面反而不一定对。于是作者进一步引入 AF2 initial guess。
这个做法的具体过程是:
- 1. 将 Rosetta 设计得到的复合物结构转换成 AlphaFold 使用的 atom positions;
- 2. 在标准 AF2 输入之外,把这些坐标作为初始结构信息传入 AlphaFold Model Runner;
- 3. 修改 AlphaFold 代码,在第一次 recycle 时,不再把 prev_pos 初始化为全零,而是初始化为输入的设计结构坐标;
- 4. 后续 recycle 由 AF2 自己根据序列、模板、pair representation 和结构模块继续更新;
- 5. 为了提升速度,作者使用减少 extra MSA 数量的配置。
这个技巧的核心不是强迫 AF2 接受 Rosetta 结构,而是给 AF2 一个合理起点,让模型判断这个复合物是否能在自身学习到的结构分布中被维持。如果 AF2 从初始设计出发仍然收敛到相似复合物,并给出低 pAE_interaction,那么这个设计更可能是真的。论文 Methods 中明确描述了这一实现细节,并提供了相应脚本。
5.5 ProteinMPNN:为什么它能提升设计效率?
AF2 过滤虽然有效,但很耗算力。论文中提到,AF2 filter 大约需要每个设计 30 GPU 秒,并且只有约 2.3% 的设计能通过,因此如果前端生成设计的效率太低,整个流程会变得昂贵。
Rosetta sequence design 在一个 minibinder backbone 上生成序列大约需要 350 CPU 秒,而 ProteinMPNN 大约只需要 2 CPU 秒。ProteinMPNN 是一个基于深度学习图模型的蛋白序列设计方法,输入蛋白骨架坐标,输出与该骨架兼容的氨基酸序列。它本质上把固定骨架条件下的序列设计变成条件概率建模问题,而不是像 Rosetta 那样在物理能量函数上进行较慢的组合优化。
作者测试后发现,在通过 AF2 pAE_interaction < 10 的候选中,ProteinMPNN 设计的实验成功率与 Rosetta 设计相近,但速度大幅提升。这说明在该流程中,慢速 Rosetta 序列优化并不是唯一可行选择。
5.6 ProteinMPNN-FastRelax:固定骨架设计与结构松弛的折中
ProteinMPNN 的一个潜在问题是,它通常在给定固定骨架上设计序列。如果输入骨架本身存在局部几何不合理或界面紧张,ProteinMPNN 可能只能在不理想的骨架上寻找序列。
因此作者提出一个混合策略 ProteinMPNN-FR:
- 1. 输入 binder-target 复合物结构;
- 2. 将 binder 序列 mask 掉;
- 3. 用 ProteinMPNN 根据复合物坐标重新设计 binder 序列;
- 4. 将新序列 thread 回 binder 骨架;
- 5. 用 Rosetta FastRelax 对复合物进行结构松弛;
- 6. 将松弛后的结构再次输入 ProteinMPNN;
- 7. 可以循环进行序列设计与结构松弛;
- 8. 最后再用 AF2 评估复合物是否通过 pAE_interaction 阈值。
这个流程保留了 ProteinMPNN 的速度优势,同时用 Rosetta FastRelax 处理骨架和侧链局部几何。论文报告 ProteinMPNN-FR 生成 pAE_interaction < 10 设计的比例约为 6.6%,吞吐量约为每 120 CPU 秒一个设计,效率达到 2.2 × 10^-6 successful designs per CPU-s equivalent;相对于原始 Rosetta design,平均每靶点效率提升约 8 倍。
5.7 前瞻性实验流程:从计算设计到湿实验验证
作者并没有只停留在回顾性分析,而是在 4 个具有生物学意义的新靶点上进行前瞻性设计:ALK、LTK、IL-10Rα 和 IL-2Rα。其中 IL-2Rα 还设计了两个不同靶位点。
实际流程大致如下:
- 1. 使用 Cao 等人的 Rosetta-based 设计流程,从靶蛋白结构和推荐 scaffold 出发进行大规模设计;
- 2. 对每个靶点生成约百万级设计并进行 AF2 预测;
- 3. 构建多个候选子集:传统 Rosetta 物理过滤组、AF2 pAE_interaction 过滤组,以及用于再设计的 AF2 复合物 RMSD 低的候选组;
- 4. 每个靶点最终选择约 2 万个候选进入实验,其中约 1.5 万个来自物理过滤,约 5000 个来自 AF2 过滤;
- 5. 总计约 8 万个设计合成 DNA,转化入酵母展示系统;
- 6. 通过靶蛋白标记、FACS 分选和深度测序估计每个设计的 SC50;
- 7. 将 SC50 < 4 μM 的设计定义为成功 binder;
- 8. 进一步选取若干 YSD 阳性设计在 E. coli 中表达纯化,并用 BLI 验证结合。
DNA library preparation 也相当工程化:设计序列被补齐到 65 个氨基酸,在 C 端加入丝氨酸 linker;序列反向翻译并针对酵母密码子优化;两端加入 adapter;寡核苷酸库经 PCR 扩增、凝胶纯化后与线性化 pETcon3 载体转化到 EBY100 酵母中。YSD 阶段先进行 avidity sort 以捕捉低亲和 binder,再用不同靶蛋白浓度进行分选,最后用深度测序估计各设计富集情况。
6. 实验设计与关键结果
6.1 回顾性结果:Type II failure 可能是更主要瓶颈
在 Type I failure 分析中,AF2/RF2 的单体预测确实能帮助识别一部分不会按设计折叠的序列。五个已有实验结构 minibinder 中,AF2 对多数 binder 单体能达到 0.2–0.8 Å Cα 精度,RF2 也表现接近。这说明对于规则、稳定的 de novo 设计小蛋白,单序列结构预测已经足够有用。
但更关键的是 Type II failure。作者发现,AF2/RF2 的复合物预测置信度,尤其是 pAE_interaction,对实验成功具有更强区分力。换句话说,很多失败设计可能不是因为 binder 自己完全折叠错了,而是因为它并不会以设计者想象的方式结合靶蛋白。
这一点很重要。它提示蛋白 binder 设计的难点不只是设计一个可折叠蛋白,而是设计一个在目标界面上具有正确相对几何、足够互补性和稳定结合倾向的复合物。
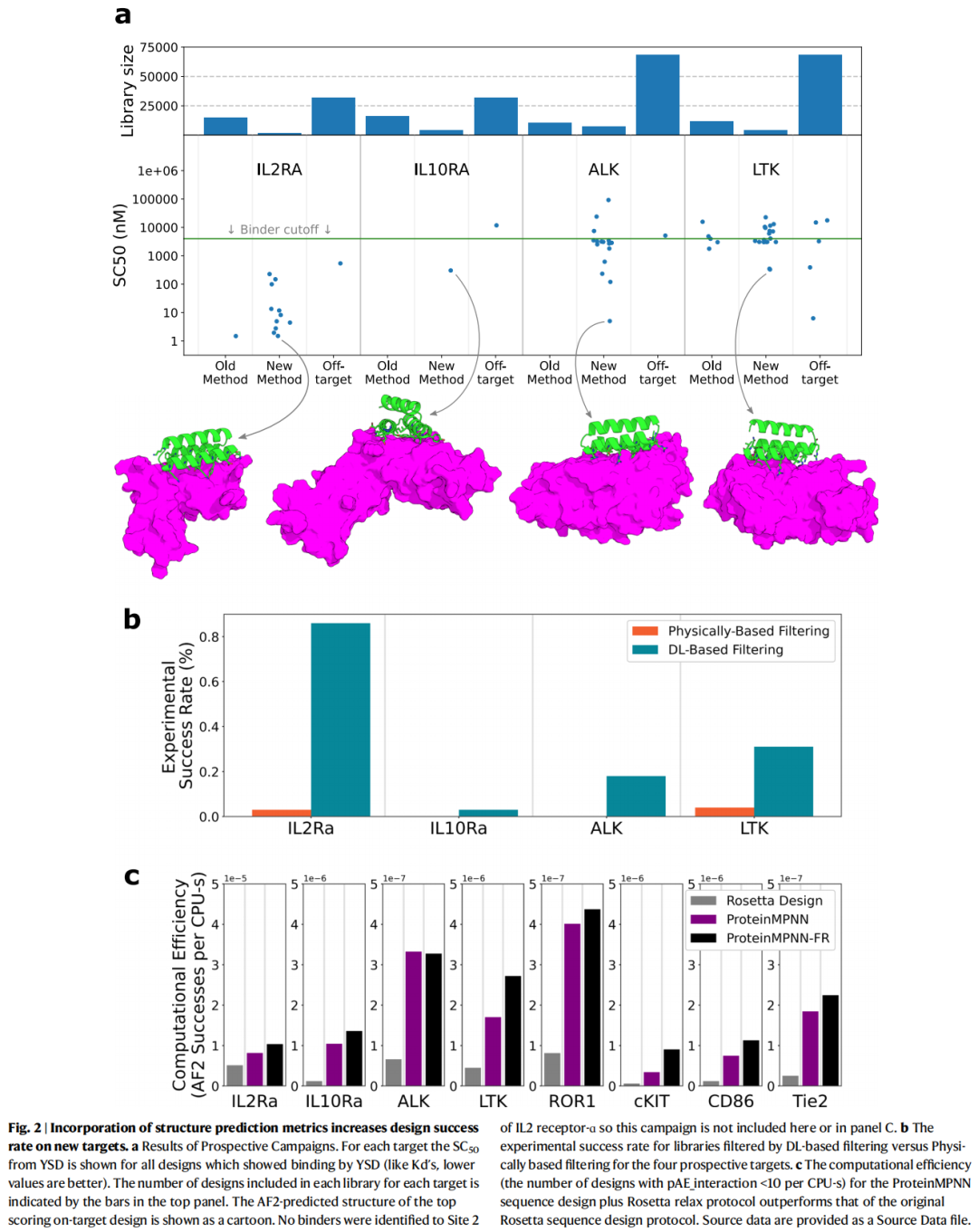
6.2 前瞻性结果:AF2 过滤显著提高实验成功率
在新靶点前瞻性测试中,AF2 过滤组在所有成功获得 binder 的靶点上都优于传统物理过滤组。
具体来看:
- • 对 LTK,AF2 过滤库相对于物理过滤库成功率提高约 8 倍;
- • 对 IL-2Rα Site 1,AF2 过滤库成功率提高约 30 倍;
- • 对 ALK 和 IL-10Rα,传统物理过滤没有得到成功 binder,而 AF2 过滤得到了成功 binder;
- • 对 IL-2Rα Site 2,两种过滤方式都没有得到成功 binder。
论文第 5 页 Figure 2 展示了不同靶点上物理过滤与 DL-based filtering 的实验成功率对比,也展示了 ProteinMPNN/ProteinMPNN-FR 在计算效率上的提升。
这些结果支持作者的核心主张:AF2/RF2 结构预测模型确实可以作为 final evaluation filter,提高进入实验的候选质量。
6.3 ProteinMPNN 结果:更快,但不牺牲成功率
作者进一步比较 ProteinMPNN 与 Rosetta 的序列设计效率。ProteinMPNN 生成序列速度远快于 Rosetta,而在经过 AF2 过滤后的实验成功率与 Rosetta 设计相近。
从效率指标看:
- • Rosetta-design 效率约为 7.6 × 10^-7;
- • ProteinMPNN design alone 效率约为 1.6 × 10^-6;
- • ProteinMPNN-FR 效率约为 2.2 × 10^-6;
- • ProteinMPNN 相比 Rosetta 平均提升约 5 倍;
- • ProteinMPNN-FR 相比 Rosetta 平均提升约 8 倍。
这里的效率定义不是单纯运行速度,而是单位计算时间内能生成多少个通过 AF2 pAE_interaction < 10 的设计。因此这个指标更接近实际设计流程中的生产能力。
启发
启发一:结构预测模型可以成为设计流程中的质量控制器
AF2/RF2 在这里不是用来预测天然蛋白结构,而是用来判断设计是否自洽。它们提供的是一种学习到的大规模结构分布先验:如果一个设计序列和设计复合物在 AF2/RF2 看来不可信,那么它进入实验的价值可能较低。
启发二:binder 设计失败要分层诊断
把失败拆成 Type I 和 Type II 是非常有启发性的。很多 AI 设计工作只报告整体成功率,但不分析失败发生在哪个环节。本文说明,只有知道失败来自折叠、界面、表达、稳定性还是实验检测,才可能针对性改进流程。
启发三:深度学习与物理建模不是简单替代关系
本文并没有完全抛弃 Rosetta。相反,它让 Rosetta 负责生成结构与物理松弛,让 AF2/RF2 负责全局结构一致性评估,让 ProteinMPNN 负责高效序列设计。这种混合路线比单一模型更符合当前蛋白设计的工程现实。
启发四:实验验证仍然是蛋白设计论文的分水岭
这篇文章的说服力不只来自回顾性 benchmark,而来自前瞻性新靶点设计、YSD 分选、深度测序和 BLI 验证。对于 binder 设计而言,湿实验命中率比漂亮的结构图更重要。
8. 局限性
8.1 成功率仍然不高
尽管作者报告接近一个数量级的提升,但绝对成功率仍然低于 1%。这意味着,即使经过 AF2 过滤,仍需大规模 DNA 合成和实验筛选。论文讨论部分也明确指出,界面能量学仍有大量未知,且 IL-2Rα Site 2 没有获得成功 binder。
8.2 初始 binder 亲和力仍有改进空间
作者指出,与原始流程类似,初始获得的 binder 多数仍处于高 nM 亲和力范围。对于治疗应用而言,往往还需要进一步亲和力成熟、特异性优化、稳定性优化、免疫原性评估和体内性质评估。
8.3 pAE_interaction 是强过滤指标,但不是结合自由能
pAE_interaction 衡量的是模型对链间相对构型的预测置信度,并不等价于真实结合自由能。低 pAE_interaction 说明 AF2 对复合物几何关系有信心,但并不保证真实热力学稳定、动力学结合快速、解离慢或在复杂生物体系中有效。
8.4 AF2 过滤可能偏向 AF2 容易相信的结构
使用 AF2 作为过滤器会带来一种潜在偏置:被保留下来的设计更像 AF2 训练分布中容易形成明确结构的复合物。这可能提升实验命中率,但也可能排除一些 AF2 不擅长处理、但真实可行的界面类型。
8.5 靶蛋白柔性和生物环境仍被简化
本文主要基于静态靶蛋白结构开展设计。真实蛋白表面可能存在构象变化、糖基化、膜环境、竞争性结合伙伴和细胞内外环境差异。对于高度柔性或构象耦合明显的靶点,静态结构设计和 AF2 过滤可能仍然不足。
8.6 特异性与 developability 评估还不完整
文章展示了 binder 可以结合目标,但对广泛 off-target 风险、长期稳定性、聚集倾向、表达产率、免疫原性和体内可开发性并没有系统展开。对于从工具分子走向药物候选,这些问题非常关键。
对 AI 制药未来发展的意义
这篇论文对 AI 制药和蛋白设计有三层意义。
第一,它说明 AI for protein design 的价值不只在生成端。很多时候,提高成功率的关键不是生成更多分子,而是在实验前更好地识别哪些候选值得合成。对于小分子生成、分子对接和虚拟筛选也是类似道理:生成能力必须与可靠过滤、排序和实验闭环结合。
第二,它推动了结构预测模型从评估工具走向设计基础设施。AF2/RF2 不再只是论文最后画结构图的工具,而可以嵌入设计流程,成为筛选、重设计和决策的一部分。
第三,它预示了后续 RFdiffusion、ProteinMPNN、BindCraft 等工作的发展方向:结构生成、序列设计、界面预测、物理松弛和实验反馈会越来越紧密地耦合。未来真正高效的 binder 设计系统,很可能不是单一大模型,而是由生成模型、结构预测模型、物理模型、实验筛选和主动学习共同组成的闭环平台。
总结与推荐理由
这篇论文不是最炫目的蛋白生成模型论文,但它非常重要,因为它回答了一个更实际的问题:当我们已经能用 Rosetta 生成大量候选 binder 时,怎样才能更有效地判断哪些设计更可能在实验中成功?
作者的答案是:先把失败拆成折叠失败和结合失败,再用 AF2/RF2 分别检查单体结构一致性和复合物界面可信度,尤其利用 pAE_interaction 过滤 Type II failure;同时用 ProteinMPNN 提升序列设计效率,用 Rosetta FastRelax 弥补固定骨架设计的局限。
推荐理由: 这篇论文清楚展示了深度学习如何以工程化方式提升 de novo 蛋白 binder 设计流程,不是单纯替代传统物理方法,而是在关键失败环节提供更有效的结构一致性判断和候选过滤。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-15,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号