Nucleic. Acids Res.| 概率扩散驱动的AI药物设计:从靶标结构感知到亲和力驱动再到保守相互作用引导

Nucleic. Acids Res.| 概率扩散驱动的AI药物设计:从靶标结构感知到亲和力驱动再到保守相互作用引导

DrugAI
发布于 2026-07-14 14:07:37
发布于 2026-07-14 14:07:37
DRUGONE
先导化合物优化连接着早期命中与临床候选,是药物发现中依赖结构洞察、化学经验与反复试验的关键环节。传统配体生成模型多从已知分子的二维表示或配体分布中学习,难以充分利用靶标口袋的三维几何、局部化学环境以及决定药效的关键残基信息。因此,生成一个“化学上成立”的分子并不等同于生成一个真正适配靶标、具有高亲和力且能形成预期相互作用的候选药物。
围绕这一核心问题,中山大学药学院雷金平副教授联合中山大学计算机学院杨跃东及王桢教授团队,以去噪概率扩散模型为统一技术底座,构建了由DiffDec、Diffleop和DiffDeCIG组成的分子设计与优化模型体系(图1)。首先,团队开发了靶标口袋三维结构感知的骨架修饰模型 DiffDec,让模型“看见口袋”,生成与口袋几何形状互补、化学环境匹配的分子;在此基础上,团队引入化学键约束和蛋白-配体亲和力引导,开发了 Diffleop,让模型“理解成键与亲和力”,将分子生成推向结构更合理、结合能力更强的化学空间;进一步地,团队通过引入氨基酸序列保守信息与蛋白-配体相互作用先验,开发了 DiffDeCIG,让模型“识别值得结合的残基”,引导生成的分子优先与功能关键的高保守位点形成相互作用。三个模型从靶标结构感知到亲和力引导再到机制导向设计,形成了一条完整的技术演进路线。
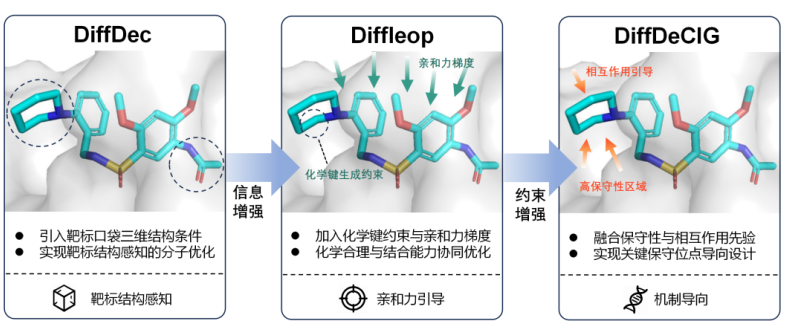
图1. 靶向性质驱动的扩散分子优化模型的递进式技术路线。
文章亮点
DiffDec:让扩散模型在三维口袋中完成靶标结构感知的骨架修饰
系列工作的第一步,是解决“生成基团能否真正适配靶标口袋”的问题。DiffDec (图2)将蛋白口袋与保留骨架作为固定条件,待生长的 R 基团原子坐标和类别在前向过程中逐步加噪,再由 E(3) 等变图神经网络在反向过程中恢复。等变结构保证模型对三维空间中的旋转和平移保持一致响应,从而能够学习口袋形状、化学环境与配体局部构象之间的对应关系。
为实现不同尺寸 R 基团的端到端生成,DiffDec 进一步设计了“虚拟原子”机制:以统一长度的原子集合承载可变规模的基团,并在去噪后移除被判定为虚拟的节点。模型既可依据用户指定的锚点进行单点或多点修饰,也能自动识别合适的锚点位置。由此,分子骨架修饰不再是脱离靶标环境的二维片段拼接,而成为受靶标口袋几何和局部化学环境共同约束的三维生成问题。
基准测试表明,DiffDec 在单点和多点骨架修饰任务中均表现出优异的生成能力。其中,单位点和多位点R基团恢复率分别达到 69.67% 和 45.34%,显著优于LibINVENT、Pocket2Mol和Flag等自回归模型。进一步的分子对接与构象分析显示,DiffDec 生成的分子不仅能够更好地占据靶标口袋,还可形成更有利的蛋白-配体相互作用,其键角和二面角分布也更接近真实分子。上述结果表明,将靶标口袋的三维结构约束直接纳入扩散生成过程,能够有效提升分子优化的准确性、结合潜力和构象合理性,为后续引入更明确的性质引导奠定了基础。
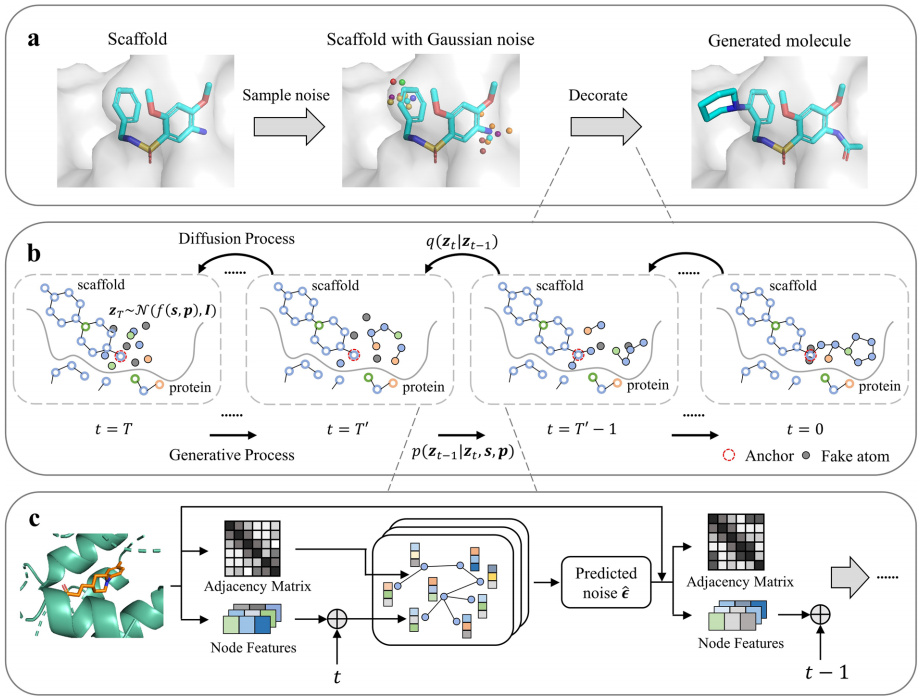
图2. 靶标结构感知的DiffDec 模型框架。
Diffleop:从口袋适配走向亲和力优化
尽管三维口袋条件能够引导分子适配靶标环境,但单纯学习口袋与配体的联合分布,并不能为提升结合亲和力提供直接的驱动力;与此同时,仅对配体原子进行扩散,也难以充分保证生成分子的成键合理性。针对这些问题,团队在靶标结构感知的基础上进一步引入化学键约束和蛋白-配体亲和力引导,开发了Diffleop模型(图3),使分子生成同时面向“结合得更强”与“结构更合理”两个目标。
Diffleop在全连接分子图上对配体原子坐标、原子类型和化学键类型进行同步扩散,并以“虚拟键”表示原本不存在的连接,使模型能够在统一的去噪过程中学习化学键的分布。与此同时,团队构建了 E(3) 等变亲和力预测网络,在每一步采样中计算亲和力相对于原子坐标、原子类型和化学键类型的梯度,并利用该梯度引导分子生成朝着亲和力提升的方向进行。这样一来,模型不再只是学习“什么样的分子可能存在”,而是在生成过程中进一步判断“怎样生成才能结合得更强”。
基准测试表明,Diffleop生成的分子在结合亲和力、蛋白-配体相互作用和配体效率等方面整体优于Pocket2Mol、AR、GraphBP、DiffLinker等模型,同时保持了合理的成键模式、局部几何构象和类药性质。消融实验进一步验证了两项引导信息的互补作用:亲和力引导决定分子向更强结合方向优化,化学键扩散则帮助模型维持结构合理性和类药性。
研究团队随后跟四川大学李国菠教授湿实验团队合作,将Diffleop用于帕金森治疗谷氨酰环化酶sQC抑制剂的优化,成功获得3个活性显著提升的候选化合物,其中最优候选物A1的 sQC 抑制活性(IC50 = 8 nM)提升了226倍、比临床试验药物PQ912高出3.5倍,很好地展示了 Diffleop模型在先导化合物优化中的应用潜力。
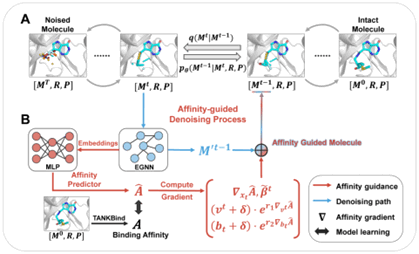
图3. 亲和力驱动的Diffleop模型框架。
DiffDeCIG:从亲和力优化走向机制引导
对于抗感染药物在内的很多药物而言,分子能否与功能关键且进化上高度保守的残基形成稳定相互作用,可能影响其作用机制、靶标选择性和潜在耐药屏障。基于此,研究团队进一步将氨基酸序列保守信息和蛋白-配体相互作用先验引入扩散生成过程,开发了 DiffDeCIG模型,使“优先与哪些残基形成相互作用”成为可被模型学习和执行的目标。
DiffDeCIG 首先通过多序列比对计算靶标序列的保守评分,并将其编码为蛋白口袋的附加特征;同时引入预训练的蛋白-配体相互作用先验网络 IPNet,学习蛋白与配体之间的关键作用模式。在扩散过程中,模型不仅学习了 R 基团原子坐标和类型的分布,还学习由相互作用先验产生的位置偏移。分子生成时,残基保守信息与相互作用先验共同调整 R 基团的生成方向,使新基团优先与高保守残基形成氢键、卤键、π-π堆积、疏水或盐桥等作用。
基准测试表明,DiffDeCIG 生成的分子能够与口袋中高保守残基形成更多非键相互作用,以提高结合潜力和生物活性。这说明与高保守氨基酸形成相互作用不再是生成后的筛选指标,而是被直接转化为模型可学习的条件信息,从而将分子生成由亲和力优化进一步推进到关键位点和特定相互作用导向的设计。
团队进一步跟中山大学周晖皓教授湿实验团队合作,将 模型应用于甲硫氨酸转运酶MetRS抑制剂的优化,并成功获得了高活性化合物 MRS-9:它对金黄色葡萄球菌 MetRS 的抑制活性 IC50 达到 0.09 μM,较初始化合物提升超过300倍,并表现出对细菌型 MetRS 的良好选择性。晶体结构研究表明,MRS-9 形成了独特的三位点占据模式,验证了保守位点与相互作用引导在提升分子靶向生物活性和选择性方面的应用潜力。
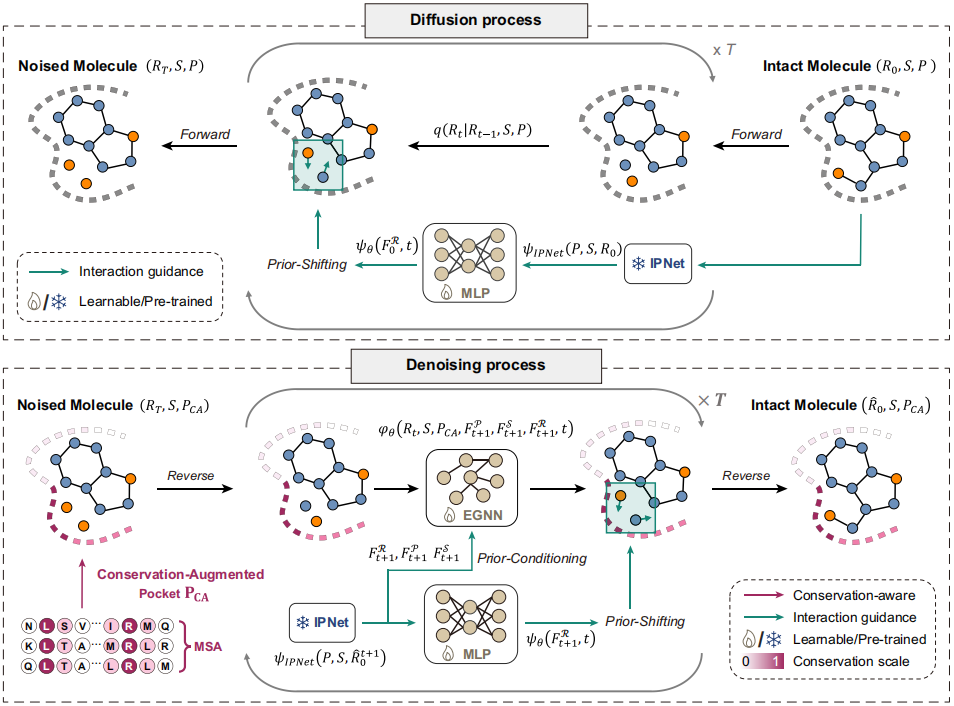
图4. 保守位点与相互作用引导的DiffDeCIG模型框架。
总结与展望
从 DiffDec 到 DiffLeop,再到 DiffDeCIG,这一系列工作展示了概率扩散用于药物分子设计与优化的清晰递进:DiffDec 以三维口袋为条件,解决“生成分子是否放得进、配得上”的结构适配问题;Diffleop加入化学键约束和亲和力梯度,进一步回答“怎样生成得更合理、结合得更强”的问题;DiffDeCIG 则叠加残基保守性和相互作用先验,开始解决“应当与哪些氨基酸结合可能更有效”的机制导向问题。模型条件由几何环境扩展到物理化学性质,再延伸至进化与功能信息,推动生成式AI药物设计从分布拟合走向多层知识约束下的目标导向优化。更重要的是,两项后续工作均实现了真实药物发现的优化任务:Diffleop获得了体外活性优于临床试验药物 的 sQC 抑制剂,DiffDeCIG 则获得了高活性、高选择性的 MetRS 三位点抑制剂,展现出从模型开发到真实药物发现的实际应用价值。
参考资料
- Su, Jingtian#; Qiao, Anjie#; Huang, Weifeng; Xu, Jingyi; Lu, Feihu; Zhang, Hao; Deng, Qirui; Zou, Jialin; Wang, Zhen*; Lei, Jinping*; Zhou, Huihao*. Discovery of a triple-site inhibitor targeting bacterial methionyl-tRNA synthetase through combined drug repurposing screening and generative AI-assisted optimization. Nucleic Acids Research, 2026, 54, gkag488.
- Anjie Qiao#, Yuting Chen#, Junjie Xie, Weifeng Huang, Hao Zhang, Qirui Deng, Jiahua Rao, Ji Deng, Fanbo Meng, Zhen Wang, Mingyuan Xu, Hongming Chen, Jiancong Xie, Shuangjia Zheng*, Yuedong Yang*, Guo-Bo Li*, Jinping Lei*. A 3D pocket-aware lead optimization model with knowledge guidance and its application for discovery of new glutaminyl cyclase inhibitors. Brief. Bioinform., 2025, 26, bbaf345.
- Junjie Xie, Sheng Chen, Jinping Lei,* Yuedong Yang.* DiffDec: Structure-Aware Scaffold Decoration with an End-to-End Diffusion Model. J. Chem. Inf. Model, 2024, 64, 2554-2564.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-13,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号