Nature | 深度网络幻觉设计:蛋白结构预测模型的另一种打开方式

Nature | 深度网络幻觉设计:蛋白结构预测模型的另一种打开方式

DrugIntel
发布于 2026-07-14 14:03:29
发布于 2026-07-14 14:03:29

论文题目: De novo protein design by deep network hallucination 期刊: Nature, 2021, Vol. 600, 547–552 作者: Ivan Anishchenko、Samuel J. Pellock、Tamuka M. Chidyausiku、David Baker 等 机构: University of Washington Institute for Protein Design、Rensselaer Polytechnic Institute 等 研究方向: AI 蛋白设计、结构预测网络反向优化、de novo protein design 核心模型: trRosetta 结构预测网络 + MCMC 序列空间优化
导读
这篇 Nature 文章提出了一个非常直接但影响深远的问题:一个训练来预测蛋白结构的深度网络,是否不仅能读懂天然蛋白,也能反过来设计从未在自然界出现过的新蛋白?作者从随机 100 个氨基酸序列出发,把序列输入 trRosetta,得到模糊的残基间距离与方向分布;随后在序列空间中进行蒙特卡洛退火优化,使网络预测的二维几何图谱逐渐变得清晰、结构化。最终,作者生成 2,000 个候选蛋白,选择 129 个进行实验表达,其中 27 个表现出与设计结构一致的圆二色谱信号,并解析了 3 个蛋白的 NMR 或晶体结构,实验结构与设计模型较好吻合。它的重要性不在于一次性解决功能蛋白设计,而在于证明结构预测网络内部确实编码了可被反向利用的序列—结构规律。
为什么这篇论文值得关注?
蛋白质设计的核心难点不是随便写出一条氨基酸序列,也不是单独生成一个看起来像蛋白的三维骨架,而是同时解决两个问题:这条序列是否会折叠?它折叠后是否就是我们设计的那个结构?
传统 de novo 蛋白设计通常依赖 Rosetta 等物理能量函数与人工设计规则。研究者先构建一个理想化蛋白骨架,再为这个骨架设计能够稳定其折叠的氨基酸序列。这一路线强在物理解释性和实验可验证性,但设计空间往往受到蓝图、拓扑、二级结构长度和人工筛选规则限制。
这篇论文提出了另一种思路:不先指定骨架,也不从天然蛋白序列改造,而是直接从随机氨基酸序列出发,反向利用结构预测网络,把随机序列逐步优化成网络认为会折叠成明确结构的序列。论文摘要中明确指出,作者从随机序列开始,经 trRosetta 预测残基间距离图,再通过蒙特卡洛采样优化预测分布与背景分布之间的 KL 散度,最终获得多种新序列与预测结构,并对其中 129 个进行实验表达和纯化验证。
这篇工作的真正价值在于,它把结构预测模型从一个给定序列、输出结构的判别式工具,转化成一个可用于生成新蛋白的能量景观或打分器。后来很多基于 AlphaFold、RoseTTAFold的蛋白设计思想,都可以在这篇工作中看到早期逻辑:既然模型学到了序列与结构之间的统计规律,就可以通过反向优化或生成式采样,把这种规律用于设计。
研究背景
一个可折叠蛋白需要同时满足多层约束。
首先是局部结构约束。氨基酸序列需要形成合理的 α 螺旋、β 折叠、turn 和 loop,主链二面角不能任意组合,疏水残基、极性残基和带电残基的位置也需要符合折叠规律。
其次是全局拓扑约束。一个 100 aa 左右的小蛋白看似不大,但残基之间存在大量长程相互作用。第 10 位残基和第 80 位残基是否接触,可能决定一个疏水核心是否形成;多个 β 链之间的配对顺序,也可能决定整个 fold 是否稳定。
第三是序列—结构耦合。给定骨架设计序列已经很难;同时设计骨架和序列更难。因为序列影响结构,结构又反过来决定什么序列合理。单独生成骨架不够,单独生成序列也不够,必须找到互相兼容的 sequence–structure pair。
在深度学习结构预测兴起之前,蛋白设计主要依赖显式物理模型和专家规则。而 trRosetta、AlphaFold 等模型的出现改变了一个事实:神经网络能够从大量天然蛋白结构和序列中学习残基间几何关系。trRosetta 原本用于预测结构,输入序列或多序列比对,输出残基对之间的距离和方向分布;如果预测图谱足够清晰,就可以通过约束最小化重建三维结构。论文明确指出,trRosetta 这类网络不仅依赖共进化信息,还可能学到了更一般的蛋白序列—结构关系。
于是,一个自然问题出现了:如果网络内部已经存储了蛋白折叠规律,能不能把网络倒过来用,让它帮助我们生成新蛋白?
以往方法
3.1 只生成序列:容易忽略明确三维结构
蛋白语言模型或序列生成模型可以学习天然蛋白序列分布,生成看起来像蛋白的序列。但序列像天然蛋白,并不等于它一定折叠成稳定、唯一、可控的结构。对 de novo 设计而言,结构可控性非常关键,尤其当我们希望设计结合蛋白、酶活性位点或特定拓扑时,仅靠序列相似性并不够。
3.2 只生成骨架:还需要解决序列编码问题
另一类方法尝试生成新的蛋白骨架,或者基于几何规则构建理想化结构。但骨架只是设计的一半。最终蛋白能否表达、溶解、折叠,取决于是否存在一条序列能稳定这个骨架。骨架生成和序列设计如果割裂,可能出现漂亮但不可编码的结构。
3.3 固定骨架设计序列:任务被简化了
RosettaDesign、ProteinMPNN 这一类思想通常解决的是 fixed-backbone sequence design:给定骨架,找一条适配骨架的序列。这非常重要,但它没有完全解决经典 de novo 设计问题,因为骨架拓扑、二级结构位置和整体形状通常已经由人或其他算法决定。
3.4 本文瞄准的缺口
这篇论文瞄准的是更原始、更开放的问题:不给定目标骨架,只给一个随机序列,能不能通过结构预测网络自身的偏好,逐步逼出一个同时合理的序列和结构?
这个问题很有意义。因为如果可行,蛋白设计就不再完全依赖人工蓝图或显式物理势能,而可以利用深度网络在训练中自动压缩得到的蛋白结构规律。
这篇论文的核心思想
这篇文章的核心思想可以概括为一句话:
把 trRosetta 看成一个可查询的蛋白结构先验,然后在序列空间中搜索,使随机序列逐渐变成网络预测为具有明确折叠结构的序列。
具体来说,随机序列输入 trRosetta 后,网络不会给出清晰结构。因为随机序列大概率不折叠,残基间距离预测是模糊的。作者的做法不是直接训练一个新生成模型,而是固定 trRosetta 参数,只优化输入序列。
优化目标也很关键。作者希望网络预测的残基间几何分布尽可能清晰,不能只是普通背景蛋白的平均状态。因此他们引入一个 background network,用来表示不依赖具体氨基酸身份的背景几何分布。然后让 trRosetta 对当前序列的预测分布尽可能偏离背景分布。偏离越大,说明网络越认为这条序列对应一个明确结构,而不是一个松散、模糊、类似 molten globule 的状态。
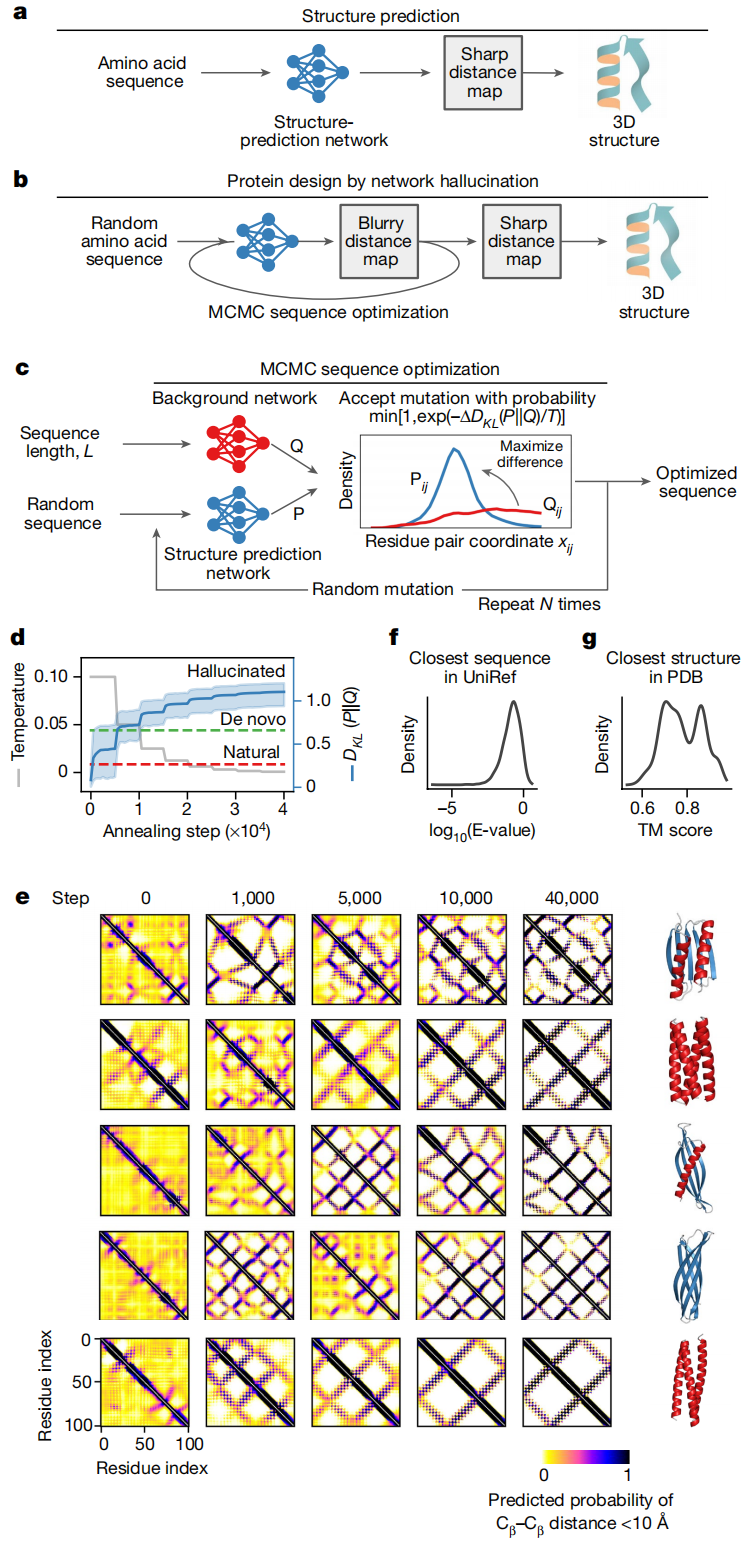

5. 方法细节:模型到底是怎么做的?
5.1 任务定义与输入输出
从概率角度看,蛋白设计可以写成寻找最大化联合概率的序列—结构对:
P(sequence, structure) = P(structure | sequence) × P(sequence)
这里有两个部分。
第一部分P(structure | sequence) 对应结构预测问题:给定序列,什么结构最可能?
第二部分P(sequence) 对应序列合理性:这条序列本身是否像蛋白序列,氨基酸组成是否过于异常,是否存在明显不利于表达或折叠的组成偏差。
本文的输入是随机生成的 100 aa 氨基酸序列。输出包括两部分:一条经过优化的新序列,以及由 trRosetta 残基间几何预测转化得到的三维结构模型。作者在整个研究中使用长度 L = 100 的蛋白,并排除了半胱氨酸,以避免在大肠杆菌还原性胞质环境中出现氧化和二硫键相关复杂性。
需要注意的是,这篇文章不是设计 binder,也不是设计酶活性位点。它设计的是可稳定折叠的新型小蛋白 scaffold。功能设计只是作者在讨论中提出的后续方向。
5.2 数据表示:不是直接生成坐标,而是生成残基间几何分布
trRosetta 的核心表示不是原子坐标,而是二维残基对几何图谱。对于蛋白中每一对残基 i、j,模型预测它们之间的距离和方向分布。
可以把它理解为一个 L × L 的二维矩阵,每个格点不是一个单值,而是一组概率分布。例如,Cβ–Cβ 距离落在哪个区间,两个残基之间的方向角落在哪个区间。方法部分说明,trRosetta 是一个二维残差卷积神经网络,输入来自单序列或多序列比对的一位点、二位点特征,输出所有残基对的距离与方向概率分布;这些 2D 预测足够准确时,可通过直接最小化转换为 3D 结构。
这种表示的优点是,它把蛋白三维折叠问题转化为残基对几何约束问题。只要距离图和方向图足够清晰,三维结构就能被约束出来。
但它也有天然局限:它主要描述主链和残基间粗粒度几何,不显式描述全原子侧链堆积、溶剂暴露、蛋白表面疏水斑块和细致能量竞争。这一点后来成为本文实验中许多蛋白形成寡聚体或聚集体的重要原因。
5.3 模型结构:两个网络,一个负责结构先验,一个负责背景参照
本文方法中有两个关键网络。
第一个是 trRosetta。它接收当前序列,输出当前序列对应的残基间几何概率分布,记作P_trRosetta。如果一条序列看起来能够折叠,trRosetta 的预测会更尖锐:某些残基对距离短、某些方向关系明确,二维图谱会出现清晰接触模式。
第二个是 background network。它的架构类似 trRosetta,也在同样训练集上训练,但不给氨基酸序列身份信息。换句话说,它只学习在给定蛋白长度、残基间序列间隔等背景因素下,普通残基对大概会有怎样的距离和方向分布。作者将其近似看成一种 generic molten globule 背景状态。
为什么需要这个背景网络?
因为单纯要求 trRosetta 输出某个结构还不够。随机序列也会输出一些概率分布,但通常很模糊。作者真正想优化的是当前序列是否让网络产生比背景更明确、更结构化的几何判断。因此目标不是让距离短就好,也不是让接触数越多越好,而是让 trRosetta 预测分布与背景分布之间的差异变大。
这就是 KL 散度进入方法的原因。
5.4 训练目标:本文不训练 trRosetta,而是优化输入序列
严格来说,本文主要不是训练一个新生成模型,而是固定已有结构预测网络,优化输入序列。目标函数包含两个部分。
第一部分是结构清晰度项:
D_KL(P_trRosetta || Q_background)
它衡量 trRosetta 对当前序列的几何预测与背景网络预测之间有多大差异。差异越大,说明当前序列越能诱导出特异、清晰的结构约束。作者对所有残基对以及距离、方向分布求平均 KL 散度。
第二部分是序列组成约束:
-D_KL(f_a || f_a^PDB)
这里f_a 是当前序列中 20 种氨基酸频率,f_a^PDB 是 PDB 中蛋白序列的氨基酸频率。这个项的作用是防止优化过程走向过于异常的氨基酸组成。否则模型可能为了制造清晰距离图,偏向某些极端序列模式,导致表达、溶解或折叠失败。
最终目标函数可以理解为:
既要让结构预测足够清晰,又不能让序列组成偏离真实蛋白太远。
方法部分明确给出了 combined objective:用 trRosetta 与 background 的 KL 散度,加上氨基酸组成相对 PDB 频率的约束,并通过模拟退火在序列空间中优化。
这个目标函数很有启发性。它不是显式最小化 Rosetta 能量,也不是直接最大化实验稳定性,而是把神经网络对可折叠性的内部判断变成了一个可搜索的目标。
5.5 生成流程:在序列空间中进行 MCMC 退火
生成流程非常具体,可以拆成以下步骤。
第一步:生成随机序列。 作者从长度为 100 的随机氨基酸序列开始,不使用天然蛋白模板,也不预设目标拓扑。
第二步:输入 trRosetta 和 background network。 对于当前序列,trRosetta 给出残基对距离和方向分布;background network 给出不依赖序列身份的背景分布。
第三步:计算目标函数。 模型计算当前序列的几何清晰度,以及氨基酸组成是否合理。
第四步:随机突变一个氨基酸。 每一步随机选择一个位置,并随机替换为另一种氨基酸。然后重新通过网络预测几何分布,重新计算目标函数。
第五步:按照 Metropolis 准则接受或拒绝突变。 如果突变让目标变好,更容易被接受;如果突变暂时变差,也可能以一定概率保留。这使搜索能够跳出局部最优。
第六步:逐步降低温度。 每条轨迹包含 40,000 次尝试突变。初始温度为 0.1,每 5,000 步减半。温度高时搜索更开放,温度低时更倾向于保留高分序列。
从视觉上看,这个过程就是让残基间距离图从模糊变清晰。随机序列开始时没有明确接触图;随着优化推进,长程接触、二级结构配对和整体拓扑逐渐浮现。原文 Fig. 1e 展示了多个轨迹中距离图随步数变清晰的过程,用于解释方法直觉。
5.6 从二维几何图谱到三维蛋白结构
优化完成后,作者并不是直接输出坐标,而是先得到一组清晰的残基间距离和方向分布。随后使用 trRosetta 的结构建模流程,通过约束最小化把这些二维几何预测转化为三维模型。
这里的关键在于:结构不是由网络直接吐出坐标,而是由网络预测的几何约束重建出来。这样做符合 trRosetta 原本的结构预测范式,也让设计出的蛋白可以用标准结构预测和结构比较工具评估。
5.7 候选筛选:从 2,000 个生成结果到 129 个实验样本
作者用上述 hallucination 流程生成了 2,000 个蛋白。接下来不是全部实验,而是进行层层筛选。
第一层是结构多样性聚类。作者使用 TM-score 比较生成结构之间的相似性,平均连接层次聚类后得到 95 个结构簇,阈值约为 TM-score 0.75。这样可以避免实验样本都集中在少数相似拓扑上。
第二层是候选打分。作者对 30 个最大簇中的设计进行评分。评分由两部分组成:一是 KL 散度,代表预测图谱相对于背景是否足够清晰;二是最终三维结构与 trRosetta 概率分布的一致性,通过交叉熵差异衡量。简单说,就是不仅要网络预测清晰,还要最终三维模型能被网络预测分布支持。
第三层是人工结构检查。作者从每个簇中选取得分最高的前 50% 或最多 20 个结构,共得到 297 个设计;随后人工剔除存在内部空腔、空洞、过大表面疏水斑块、二级结构形成不良等问题的模型。3 个簇因模型质量差被完全剔除。最终从剩余 27 个结构簇中选择 129 条序列做实验验证,每个簇不超过 10 个设计。
这一步非常重要,也很容易被忽略。本文不是完全端到端地从模型输出直接进实验,中间仍然有打分、聚类、人工检查和实验可行性筛选。这说明当时的网络幻觉设计已经展示了生成能力,但要进入实验仍需要较强的设计经验和过滤流程。
5.8 实验验证流程:从计算设计走向真实蛋白
作者对 129 个候选蛋白进行了湿实验验证,流程包括:
- 1. 合成编码这些设计蛋白的基因;
- 2. 克隆到 pET28b(+) 表达载体,并加入 N 端 His-tag 和 thrombin cleavage site 便于纯化;
- 3. 在 E. coli BL21(DE3) 中表达;
- 4. 用镍柱亲和层析进行初步纯化;
- 5. 用 SEC 检查蛋白是否形成单体或小寡聚体;
- 6. 用 CD 光谱判断二级结构是否符合设计;
- 7. 对表现较好的蛋白进行 NMR、SEC-MALS 或 X-ray 晶体学结构解析。
这套验证链条的价值在于,它不只看计算模型得分,也不只看是否表达,而是逐层检查:蛋白是否可溶、是否单分散、是否具有预期二级结构、是否稳定、最终三维结构是否与设计模型一致。
6. 实验设计与关键结果
6.1 生成结果是否多样?
作者生成 2,000 个 hallucinated proteins,并分析其序列空间和结构空间。结果显示,不同 MCMC 起点会收敛到不同的序列—结构对。生成结构覆盖 all-α、all-β 和 mixed α–β 等类别,并在 TM-score 0.75 聚类阈值下得到 95 个 sub-fold。原文 Fig. 2 展示了序列空间、结构空间以及代表性拓扑。
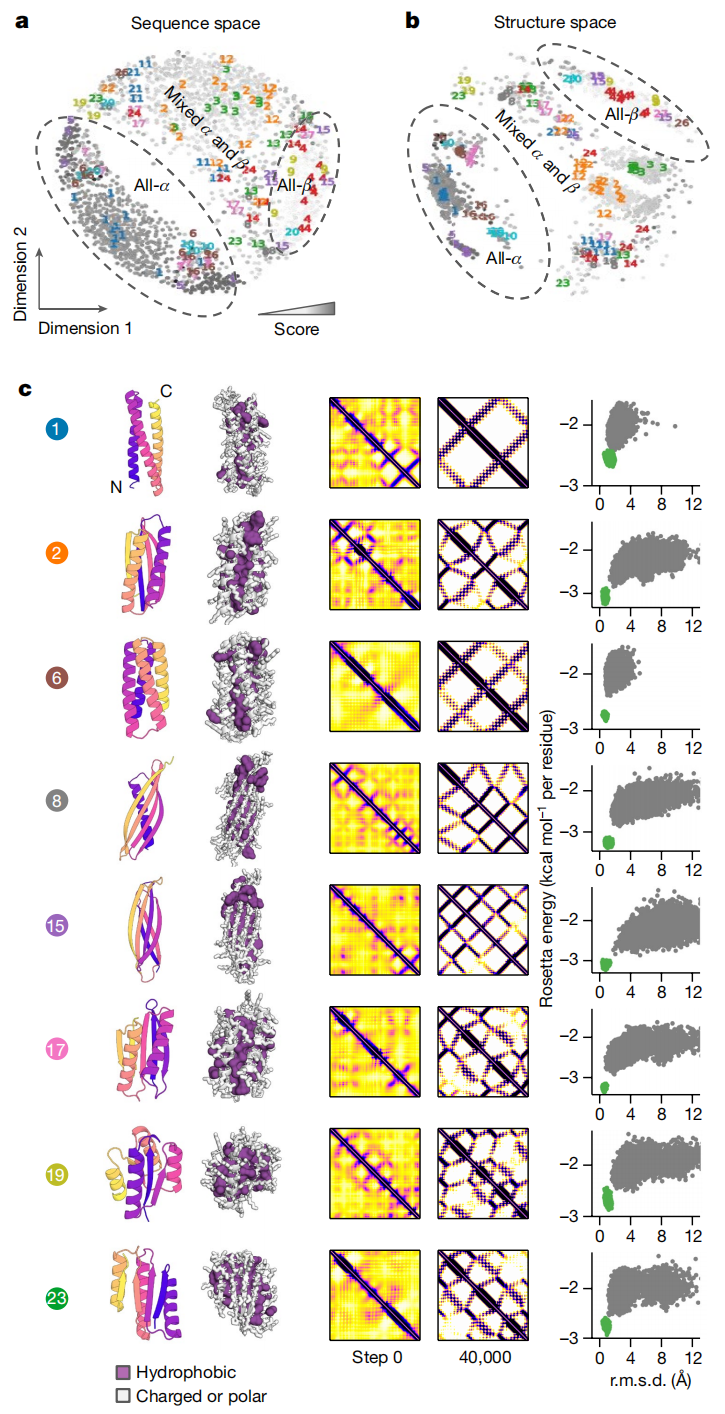

这说明模型不是只找到一个固定解,也不是简单复制某个天然蛋白,而是在网络先验允许的范围内产生了多种拓扑方案。
当然,多样性并不等于完全新颖。作者也指出,这些结构往往更像理想化 de novo 设计蛋白,而不是天然蛋白。它们具有规则 α 螺旋、β 折叠和较短 loop,缺少天然蛋白中常见的复杂插入和不规则区域。这个结果很有意思:网络训练于天然蛋白,但反向优化后却偏向更理想、更规则、更设计化的结构。
6.2 Rosetta 是否认为这些序列能折叠到目标结构?
为了进行独立计算验证,作者使用 Rosetta ab initio folding simulations 检查 hallucinated sequence 是否会在 Rosetta 能量函数下折叠到 hallucinated structure。这里的独立性很重要:trRosetta 用于生成,Rosetta 能量函数没有参与序列生成或实验样本选择。论文显示,在 129 个实验候选中,82 个 Rosetta 最低能结构与对应 hallucinated structure 的 Cα RMSD 小于 3.0 Å;并且对全部 129 个蛋白,从设计模型出发采样得到的最低能结构能量低于从延展链出发得到的其他结构。
这个结果说明,至少从 Rosetta 物理能量模型看,很多网络生成的序列确实编码了对应结构。这是对纯神经网络目标函数的一个重要补充。
6.3 实验表达和 CD 验证结果如何?
作者合成了 129 个设计蛋白并在大肠杆菌中表达纯化。结果中有 27 个蛋白在 SEC 中呈现单体或小寡聚体峰,并继续进行 CD 光谱验证;这些蛋白的 CD 光谱与目标二级结构一致。21 个蛋白表现出较高热稳定性,表观熔解温度超过 70 °C;其中部分 mixed α–β 设计在 95 °C 之前没有明显解折叠。
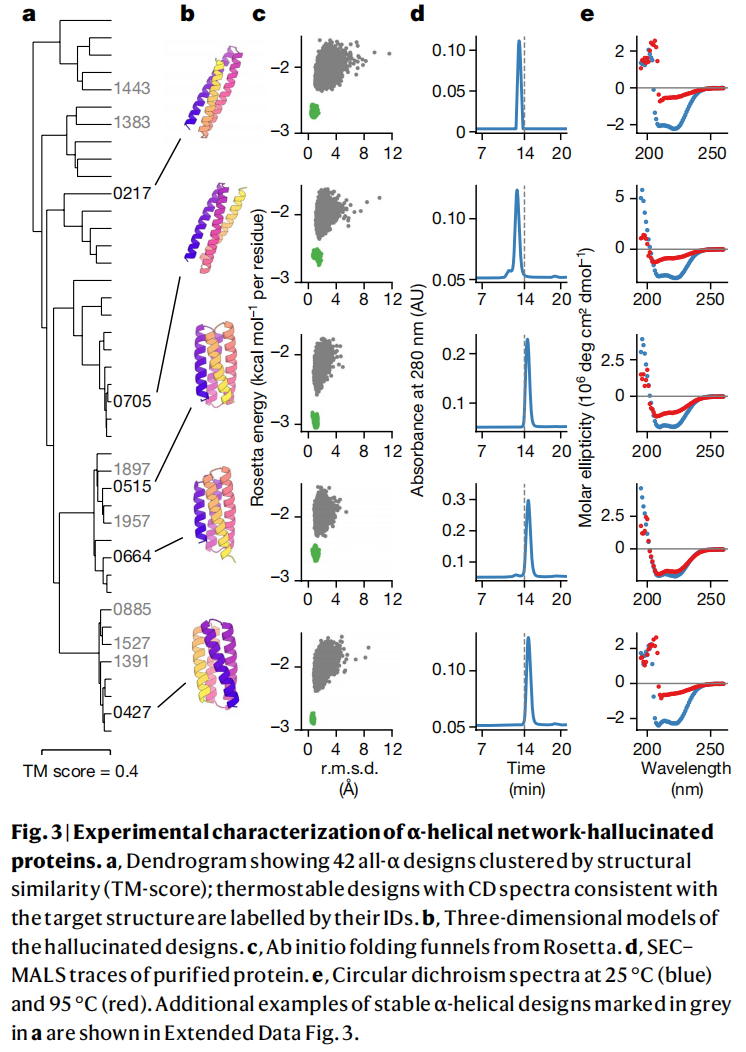
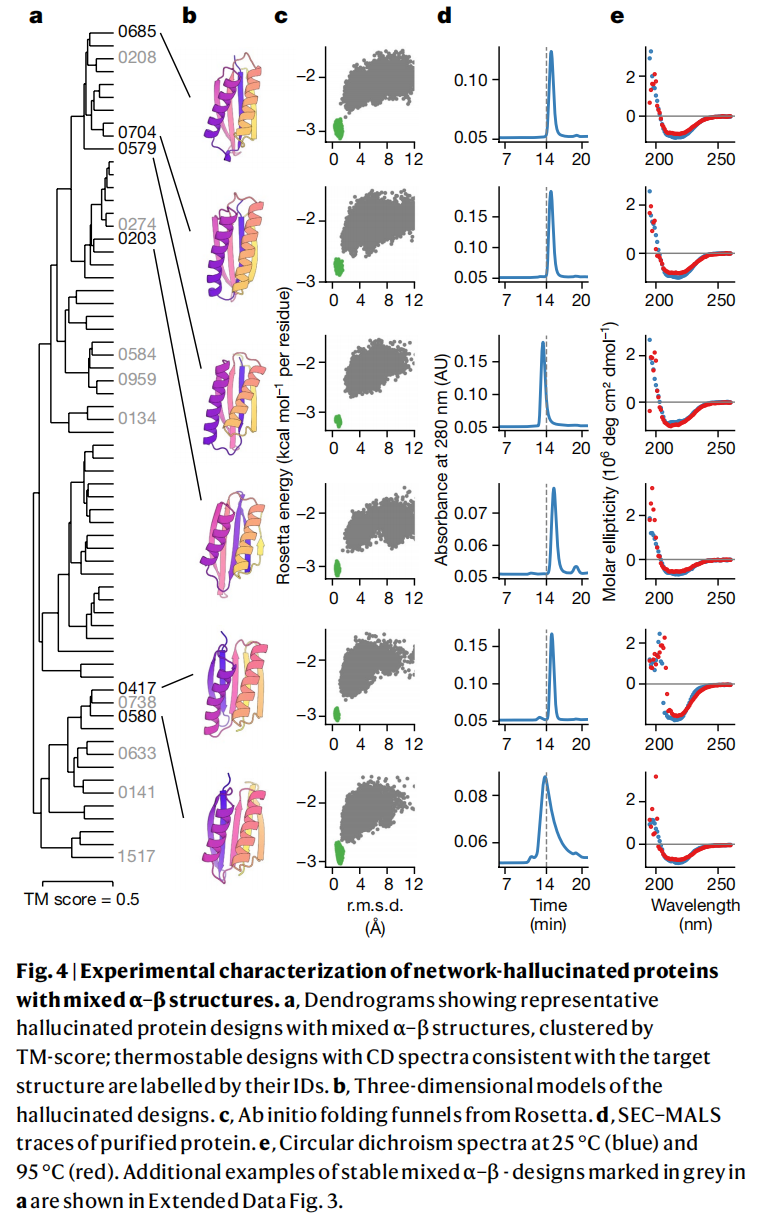
这个结果说明,网络幻觉生成并不只是计算上的图形游戏。至少有一部分设计蛋白可以真实表达、纯化,并展现预期二级结构和稳定性。
不过也要注意成功率的解释。129 个进入实验的候选已经经历了计算筛选和人工过滤,其中 27 个进入更深入表征,约占 21%。这个比例对于早期 de novo 蛋白设计并不低,但也说明方法还不是按下按钮即可大规模产生高成功率单体蛋白。
6.4 三维结构是否真的吻合?
这篇论文最有说服力的证据来自结构解析。
设计 0515 的 NMR 结构是一个单体反平行四螺旋束,20 个结构集合与 hallucinated model 的 Cα RMSD 约为 1.82 Å。
设计 0217 的晶体结构分辨率为 2.9 Å,是一个三螺旋束,与 hallucinated model 整体折叠相似,100 个残基上的主链 RMSD 为 2.53 Å。
设计 0738 经过表面疏水残基极性替换得到 0738_mod,并解析出 2.4 Å 晶体结构。整体与原 hallucinated model 的 Cα RMSD 为 3.68 Å;尽管存在配准偏移,N 端和 C 端局部区域分别以 1.32 Å 和 2.17 Å RMSD 与模型吻合。原文 Fig. 5 集中展示了这些设计模型与实验结构的叠合结果。
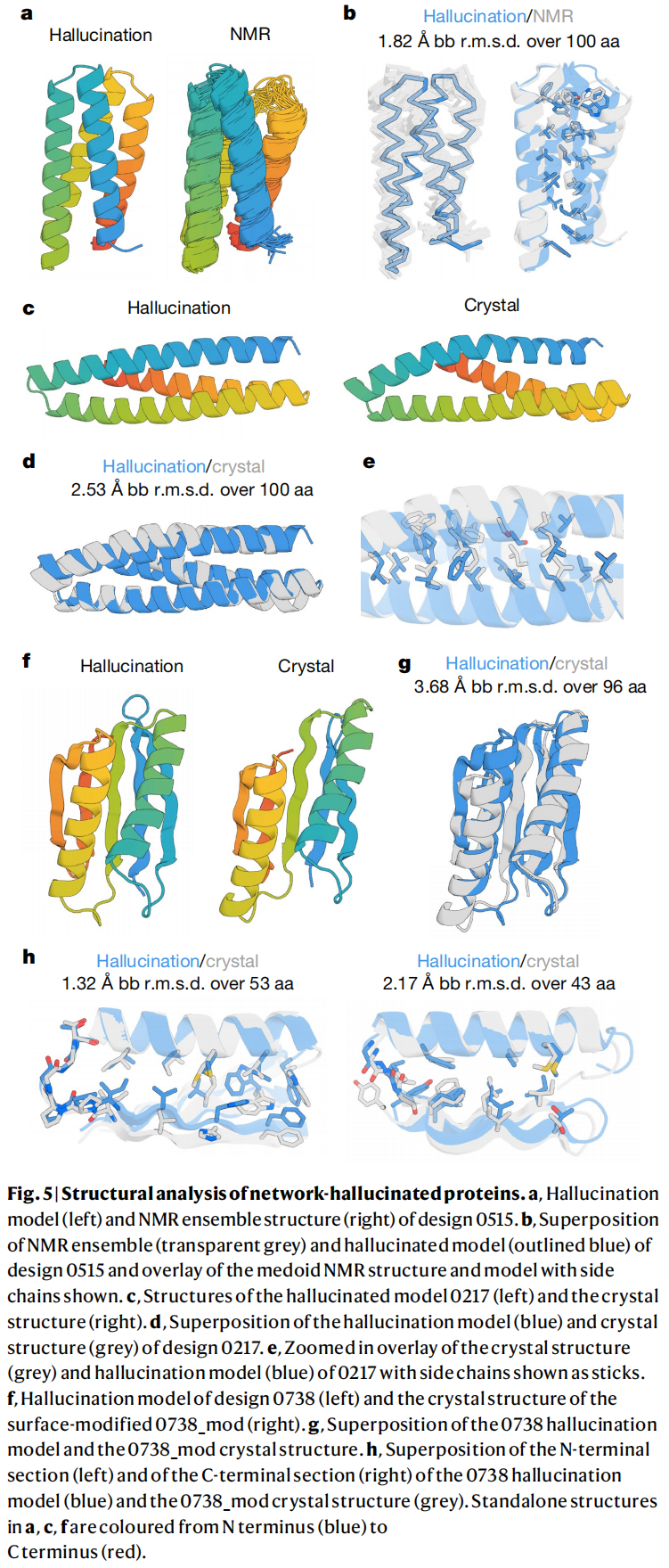
这一部分是论文最关键的证据链。因为 CD 只能证明二级结构大体一致,而 NMR 和晶体结构能证明三维折叠确实接近设计模型。对于一个从随机序列开始、没有显式物理折叠知识的神经网络反向优化方法来说,这已经是非常强的验证。
启发
启发一:结构预测网络内部学到的不只是共进化信号
trRosetta 原本很大程度上依赖多序列比对和共进化信息来预测天然蛋白结构。但本文从单条随机序列出发,通过优化序列让网络产生明确结构,说明网络参数中确实编码了某些更一般的蛋白几何规律。
这不等于网络完全理解了蛋白物理,但说明它学到的统计先验足够强,可以被反向利用。
启发二:生成模型不一定要从头训练
本文没有重新训练一个专门的蛋白生成模型,而是把已有预测网络当作可微或可查询的评价器,通过输入优化完成设计。这种思想后来在很多 AI for Science 任务中反复出现:当一个预测器足够强时,可以通过反向优化、梯度引导、采样搜索或能量重排,把预测器转化成设计工具。
启发三:清晰的中间表示非常关键
本文没有直接优化黑箱结构坐标,而是优化残基间距离与方向分布。这个中间表示具有很强结构含义:清晰的距离图代表明确折叠,模糊的距离图代表未定义结构。这使得目标函数比单纯的网络置信度更可解释。
启发四:AI 蛋白设计仍然需要物理和实验闭环
这篇工作虽然展示了神经网络生成能力,但最终可信度来自 Rosetta、SEC、CD、NMR 和 X-ray 的多层验证。没有这些验证,hallucinated proteins 仍可能只是网络喜欢的模式。AI 负责提出候选,物理与实验负责确认候选是否真实存在。
对 AI 制药和蛋白设计未来发展的意义
1 预测模型可以成为设计模型
这篇文章最重要的思想是:强预测模型可以被反向使用。对于 AI 制药,这不仅适用于蛋白结构,也适用于分子性质预测、蛋白—配体相互作用预测、抗体亲和力预测和 ADMET 预测。只要预测器足够可信,就可以作为设计目标的一部分;但前提是必须处理好分布外泛化和目标函数作弊问题。
2 蛋白设计会从无约束生成走向条件控制
本文从随机序列生成可折叠 scaffold。作者在讨论中提出,未来可以把目标函数扩展到特定结构特征,例如 binding motifs 或 catalytic sites,让网络围绕功能位点 hallucinate 支撑骨架。论文也提到该方法可以扩展到 RoseTTAFold 和 AlphaFold2,并可通过梯度回传提高采样效率。
这正是后续蛋白设计发展的关键方向:不是只生成能折叠的蛋白,而是围绕功能位点、结合界面、抗原表位或催化几何进行条件设计。
3 AI 设计需要全原子物理补偿
本文暴露出的主要问题,如表面疏水斑块、寡聚、侧链 packing 不充分,提示单靠粗粒度结构预测网络还不够。未来更可靠的系统需要把深度网络的全局结构先验与全原子能量、侧链设计、溶剂暴露、界面能和实验反馈结合起来。
4 实验闭环仍是 AI 蛋白设计的最终评判标准
这篇论文之所以有说服力,不是因为模型图谱很好看,而是因为作者做了表达、纯化、CD、NMR 和晶体学验证。对于 AI 制药来说,这一点尤其重要。任何生成式模型如果只在计算指标上自我验证,很容易被模型偏好或 benchmark 偏差误导。真正能推进领域的设计,需要把生成、筛选、合成、表征、结构解析和功能测试连成闭环。
推荐理由: 这篇文章是理解 AI 蛋白设计从结构预测走向生成设计的重要节点,尤其适合读者理解预测网络反向优化、序列—结构联合设计、实验验证闭环以及后续 RFdiffusion/ProteinMPNN 类工作的思想来源。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-13,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号