万亿级化学空间导航:早期药物发现的范式转变

万亿级化学空间导航:早期药物发现的范式转变

DrugIntel
发布于 2026-07-03 20:55:24
发布于 2026-07-03 20:55:24

文献来源:Korn M, Ehrt C, Ruggiu F, Gastreich M, Rarey M. Navigating large chemical spaces in early-phase drug discovery. Current Opinion in Structural Biology, 2023, 80: 102578. DOI:10.1016/j.sbi.2023.102578 发表时间:2023 年 4 月 4 日
目录
- 1. 背景与研究动机
- 2. 化学空间的生成方法
- 3. 基于分子相似性的搜索策略
- 4. 基于蛋白结构的化学空间搜索
- 5. DNA 编码库技术(DELs)
- 6. 商业化学空间与产业应用
- 7. 工业界的实际部署案例
- 8. 技术局限性与未来展望
- 9. 绿色计算与可持续性思考
一、背景与研究动机
1.1 传统范式的困境
早期药物发现的核心任务是:在庞大的化学宇宙中高效定位具有生物活性、满足多重药理学标准的小分子候选药物。
在传统模式下,高通量筛选(High-Throughput Screening, HTS)——无论是实验性 HTS 还是虚拟 HTS——是寻找先导化合物的主流手段。然而,这一范式面临根本性瓶颈:
- • 覆盖范围有限:商业实体化合物库通常仅有数百万至数千万个化合物,而理论可合成的类药分子空间估计超过 10⁶⁰ 量级;
- • 成本高昂:实体化合物库的维护、自动化筛选平台的运营以及后续的化学合成均需巨大投入;
- • 知识产权瓶颈:大量已筛选化合物与已知专利高度重叠,新颖 IP 的获取愈发困难;
- • 顺序筛选效率低下:对于十亿量级以上的化合物集合,逐一处理的顺序算法在计算资源和时间上均不可行。
1.2 新兴范式的崛起
近年来,三种技术路线的汇聚正在打破上述范式限制,推动早期药物发现进入大化学空间导航时代:
技术路线 | 核心优势 | 代表规模 |
|---|---|---|
按需合成的组合化学空间(Make-on-Demand Spaces) | 高合成成功率、低成本、可购买 | 10⁸ ~ 10¹² |
生成式机器学习(Generative ML) | 探索前所未见的分子多样性 | 理论无限 |
DNA 编码库(DELs) | 实验端亿级化合物同步筛选 | 10⁸ ~ 10¹² |
这三种技术相互独立又彼此互补,共同构成了新一代早期药物发现的计算与实验平台。
1.3 计算化学信息学的核心挑战
规模的爆炸性增长对计算方法提出了全新要求。作者将核心计算问题归结为两个几乎正交的子问题:
- 1. 如何定义和构建搜索空间(How to shape the search space)
- 2. 如何在空间中高效搜索(How to search in it)
传统的顺序处理(sequential processing)算法在十亿量级以上已完全失效,这驱动了一系列基于组合优化和机器学习的新型化学信息学工具的快速发展。
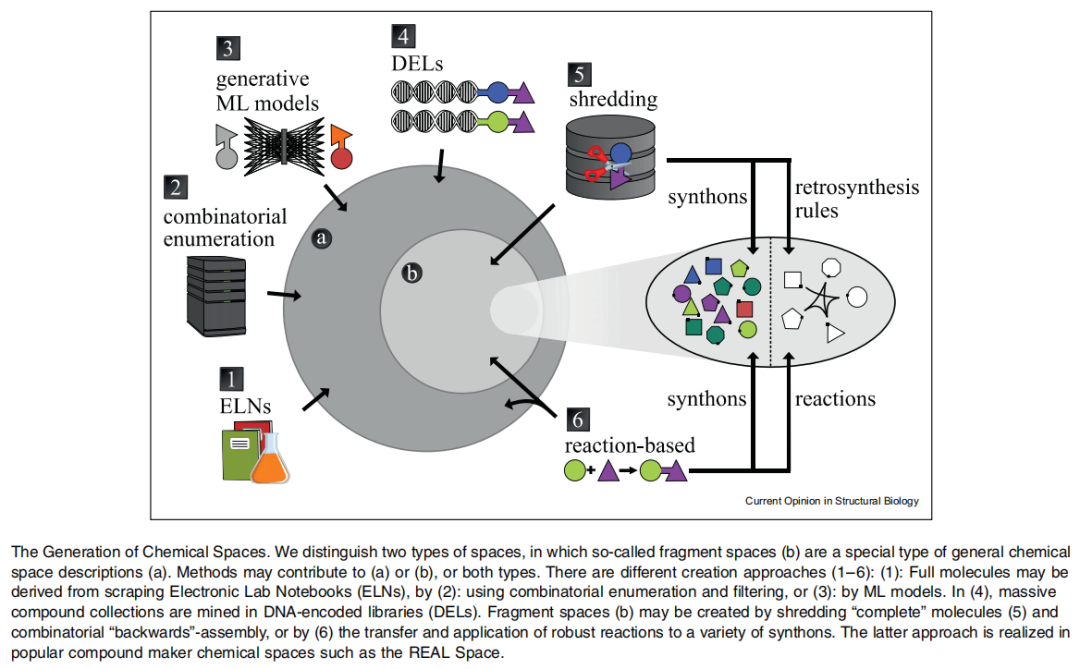
二、化学空间的生成方法
2.1 全枚举方法(Full Enumeration)
最直接的化学空间生成方式是对所有可能的有机小分子进行系统性枚举。以 GDB-17(化学宇宙数据库)为代表,该工作对最多含 17 个重原子的所有有机分子进行了枚举,得到约 1,660 亿个结构。
然而,全枚举方法的根本局限在于:生成的大量化合物合成难度极大,无法满足早期药物发现对"可合成性"的核心需求。
2.2 基于合成规则的片段化学空间(Fragment Spaces)
这是目前最具实用价值的化学空间生成策略,其核心思路是:
用合成反应规则 + 构建块(building blocks/synthons)来隐式描述化学空间,而非显式枚举所有化合物。
主要技术路线:
(1)逆合成切割与重组(Retrosynthetic Fragmentation)
- • 代表方法:RECAP(Retrosynthetic Combinatorial Analysis Procedure),将已知药物分子按合成规则切割为片段,再重新组合产生新分子;
- • 后续发展:FlexNovo、DOGS 等工具进一步整合了三维结构匹配与de novo设计能力。
(2)基于稳健反应的正向合成设计
- • Hartenfeller 等人整理了一套用于计算机辅助分子设计的稳健有机合成反应集合(约 58 种);
- • 将这些反应与大量兼容构建块配对,即可生成万亿量级的理论化合物空间;
- • 关键突破:这类空间无需存储枚举形式,仅用"反应配方"描述,普通台式机即可处理。
(3)开源工具支持
- • OpenChemLib(Wahl & Sander, 2022):全自动化创建虚拟化学片段空间;
- • SynthI(Zabolotna et al., 2022):基于合成子的文库设计新工具,强调化学空间的合理设计而非单纯搜索。
化学空间的两种形式:
一般化学空间(General Chemical Space)
├── 包含完整分子(来自 ELN 抓取、枚举过滤、ML 生成)
└── 片段化学空间(Fragment Space)← 特殊子类
├── 由"切碎"完整分子(逆合成拆分)生成
└── 由反应规则正向应用于合成子生成(如 REAL Space)2.3 生成式机器学习方法
生成式 ML 代表了一种完全不同的化学空间探索哲学——无需显式定义化学空间,直接生成满足期望性质的分子。
主要模型架构:
模型类型 | 代表架构 | 特点 |
|---|---|---|
循环神经网络 | RNN(SMILES 生成) | 训练简单,但生成分子多样性有限 |
生成对抗网络 | GAN | 可生成高多样性分子,训练稳定性差 |
变分自编码器 | VAE | 连续潜空间,支持属性导向优化 |
扩散模型(新兴) | 图扩散、3D 扩散 | 可直接生成三维构象 |
特别值得关注的进展:
- • Méndez-Lucio 等(2020)将生成模型与基因表达谱直接挂钩,可根据细胞水平的表型数据生成候选分子,突破了传统靶点驱动设计的局限;
- • Dolfus 等(2022)提出了合成路线感知的结构类似物生成方法,直接在合成路径层面操控分子生成,大幅提升可合成性。
核心局限:生成式 ML 产生的分子往往合成可行性未知,需配合合成可行性预测模型(如 Gao & Coley, 2020)进行过滤,或直接训练合成感知生成模型。
2.4 DNA 编码库(实验端空间生成)
DELs 可视为实验端的"化学空间生成与搜索一体化"技术,详见第五节。
三、基于分子相似性的搜索策略
3.1 传统方法的局限
分子相似性虚拟筛选是计算药物发现中最经典的工具,主要依赖:
- • 拓扑指纹(如 ECFP、MACCS)的 Tanimoto 相似度;
- • 最大公共子结构(MCS)搜索。
对于十亿量级以下的数据库,顺序搜索算法仍可接受。但面对片段空间中的 10¹²~10¹⁵ 量级,传统方法彻底失效。
3.2 组合算法的突破
(1)FTrees-FS(2001)——第一个精确组合搜索算法
- • 基于"约化图相似度"(Reduced Graph Similarity)概念;
- • 通过将分子用反应和反应物的片段空间隐式表示,实现次线性时间(sublinear time)内的最相似分子提取;
- • 意义:证明了组合化学空间的精确搜索在计算上是可行的。
(2)CATS(Chemically Advanced Template Search)
- • 利用包含原子对距离、电荷、亲脂性和供/受体性质的向量表示的欧氏距离进行相似性搜索;
- • 核心优势在于捕捉药效团层面的相似性,支持骨架跃迁(scaffold hopping)。
(3)SpaceLight(2021)——指纹相似性搜索的组合算法
- • 专为大型组合片段空间设计;
- • 在次线性时间内完成基于分子指纹的相似性搜索;
- • 发表于 J. Chem. Inf. Model.(Bellmann et al., 2021),被列为本综述的"特别关注"文献。
(4)SpaceMACS(2022)——最大公共子结构的精确搜索
- • 首个能在含 10¹⁵ 个分子的化学空间中执行精确结构搜索的算法;
- • 基于组合思维,将片段结果组合为完整分子的搜索结果;
- • 这是结构搜索领域的里程碑式成果。
(5)SmallWorld(NextMove Software)
- • 商业软件,同样支持超大规模数据库的组合搜索;
- • 在工业界获得广泛应用,但算法细节尚未在学术文献中详细披露。
3.3 三维形状搜索
基于分子形状叠合的虚拟筛选对于发现具有相同蛋白结合模式但拓扑结构差异较大的化合物尤为重要:
- • FastROCS(OpenEye)和 RIDE(Molsoft):GPU 加速的形状叠合实现,可处理数百亿化合物,但需要大量硬件资源;
- • Galileo(Meyenburg et al., 2023):首个面向大型组合片段空间的基于药效团的三维搜索方法,采用启发式策略。
三维搜索的根本挑战: 与拓扑搜索不同,三维搜索通常不满足组合优化所需的"局部解可组合为全局解"的条件(partial solution independence),因此精确组合算法的设计极为困难。作者判断:在三维搜索领域,启发式方法将在未来数年内持续主导。
四、基于蛋白结构的化学空间搜索
4.1 分子对接的挑战与机遇
当靶蛋白的实验结构或高质量同源模型可用时,分子对接(molecular docking)是探索大化学空间中真正新颖生物活性分子的最无偏差方法。其流程为:
- 1. 在结合位点内生成分子姿态(poses);
- 2. 用打分函数评估蛋白-配体相互作用;
- 3. 对候选分子排序,优先合成/购买排名靠前的化合物。
然而,传统对接工作流的根本瓶颈是:对于十亿~百亿量级的文库,逐一对接在运行时间和硬件资源上完全不可行。
4.2 从头设计方法的演化
组合思维在基于结构的药物设计(SBDD)中有悠久历史,关键进展包括:
方法/工具 | 年份 | 策略 |
|---|---|---|
LUDI | 1992 | 基于片段的酶抑制剂从头设计 |
GroupBuild | 1993 | 片段叠加与生长 |
FlexNovo | 2006 | 大型片段空间中的结构搜索 |
ALTA-VS | 2006 | β-分泌酶抑制剂的计算发现 |
历史局限:上述方法生成的分子往往合成可行性极差,大幅限制了实际应用价值。
4.3 新一代化学空间对接方法
随着可合成片段空间的成熟,结构导向的化学空间搜索进入新阶段:
(1)片段延伸策略(Fragment Extension)
代表工作:
- • Synthon-based Docking(Sadybekov et al., 2022, Nature):将对接算法与组合化学底层逻辑深度整合,实现对非枚举的百亿级化学空间的对接。该工作不仅发现了亚微摩尔级活性化合物(CB2 受体抑制剂),更证明了算法的可扩展性——★★ 本综述最高推荐文献之一;
- • FastGrow(Penner et al., 2022):实时片段生长工具,已应用于 DYRK1A 抑制剂设计;
- • Chemical Space Docking(Beroza et al., 2022, Nat. Commun.):发现 ROCK1 激酶抑制剂的结构导向化学空间搜索。
(2)"晶体结构优先"策略(Crystal Structure First)
Müller 等(2022)提出的 "Magnet for the Needle in Haystack" 策略:
- • 先通过实验 X 射线片段筛选获取弱结合片段;
- • 再以这些片段为锚点,计算延伸至十亿级组合空间中高亲和力的可购买分子;
- • 结果:所获分子高亲和力且完全可购买,极大缩短了从苗头化合物到先导化合物的时间。★★ 本综述最高推荐文献之一
(3)进化搜索策略
Galileo 和系统进化化学空间探索(Lu et al., 2022)将进化算法引入化学空间搜索,通过迭代优化在三维空间中高效寻找活性分子。
4.4 关键基准工作
Penner 等(2020)建立了贴近真实交叉对接场景的评估基准,为上述方法的系统性比较提供了标准框架,推动了该领域的良性发展。
五、DNA 编码库技术(DELs)
5.1 技术原理
DNA 编码库(DNA-Encoded Libraries)技术由 Brenner & Lerner 于 1992 年提出,其核心是:
将每个化合物用唯一的 DNA/RNA 序列标记,从而实现亿级化合物的并行合成与筛选。
标准筛选流程(Panning):
DEL 文库
↓ 与固定化靶蛋白孵育
结合物 + 非结合物混合物
↓ 洗涤去除非结合物
富集的结合物(DNA 标签完整)
↓ DNA 高通量测序
命中物的化学结构解码
↓ 频率统计 + 聚类分析
苗头化合物家族鉴定信号去噪关键:
- • 引入对照(off-target pan)检测非特异性结合;
- • 通过频率计算和富集倍数分析过滤假阳性;
- • 结合聚类/可视化识别化学型家族。
5.2 DEL 的独特优势
- • 规模:单次实验可同时探索 10⁸ ~ 10¹² 个化合物;
- • 密度:组合性质使 DEL 能比 HTS 更密集地覆盖特定化学空间区域;
- • 多样性:涵盖共价结合物、PROTAC 分子、片段等多种类型;
- • 绿色化学:用量极小(皮克至纳克级),大幅减少化学废弃物。
5.3 机器学习与 DEL 数据的协同
DEL 技术与 ML 的深度整合正在开辟新范式:
(1)分类模型——从 DEL 数据构建虚拟筛选模型 McCloskey 等(2020, J. Med. Chem.)发表了 DEL 数据训练 ML 分类模型的首个成功案例:
- • 利用含噪声的 DEL 数据训练分类模型;
- • 在三个前瞻性虚拟筛选中成功发现新化学实体;
- • 意义:即使命中物本身并非新 IP,ML 模型仍可预测 IP 空间之外的新结构。★★ 本综述最高推荐文献
(2)回归模型——结合亲和力排序预测 Ma 等(2021, NeurIPS AI4Science Workshop)展示了 DEL 数据上的回归 ML 模型:
- • 有效去噪,并预测新化合物的结合亲和力排序;
- • 可防止合成无活性化合物,加速苗头到先导的优化过程。★ 特别关注
(3)数据去噪的方法学进展
- • 引入协变量(非特异性结合、标签起始群体不平衡)的 ML 去噪;
- • 处理"部分产物"(partial products)问题(Binder et al., 2022)。
5.4 技术局限与突破方向
现有局限:
- • DEL 兼容的合成反应种类有限(须在 DNA 存在条件下进行),化学多样性受约束;
- • 通常需要固定化靶蛋白,对膜蛋白和非可溶靶标适用性差;
- • 命中率去噪和化学型解析仍有较高假阳性率。
进展方向:
- • DEL 兼容反应空间持续扩展;
- • 非固定化靶标(溶液相)、膜蛋白、细胞裂解液筛选方法逐步成熟;
- • 靶标兼容性范围正在快速拓展。
六、商业化学空间与产业应用
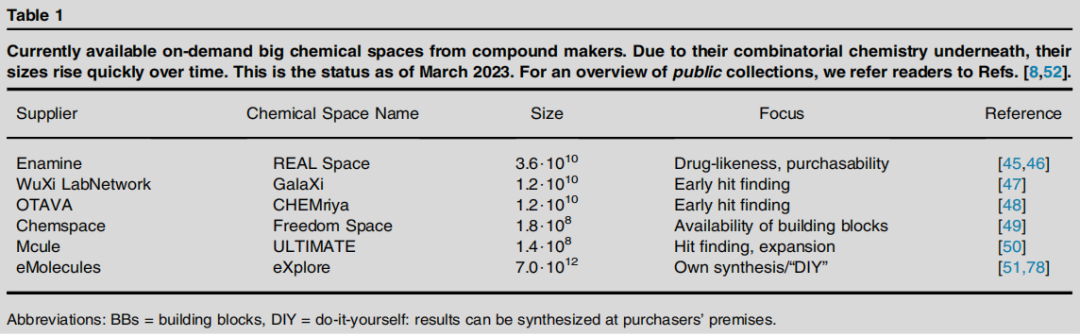
6.1 商业按需化学空间现状(2023 年 3 月)
供应商 | 化学空间名称 | 规模 | 定位 |
|---|---|---|---|
Enamine | REAL Space | ~3.6 × 10¹⁰ | 类药性、可购买性 |
WuXi LabNetwork | GalaXi | ~1.2 × 10¹⁰ | 早期苗头发现 |
OTAVA | CHEMriya | ~1.2 × 10¹⁰ | 早期苗头发现 |
eMolecules | eXplore | ~7.0 × 10¹² | 自合成(DIY) |
Chemspace | Freedom Space | ~1.8 × 10⁸ | 构建块可用性 |
Mcule | ULTIMATE | ~1.4 × 10⁸ | 苗头发现与扩展 |
核心价值:
- • Enamine REAL 体系(乌克兰公司)的突破性意义:将化合物合成成本降低一个数量级,且实际项目中合成成功率超过 85%;
- • 各商业空间的分子重叠度出人意料地非常低(Bellmann et al., 2022),说明它们在化学空间中各有侧重,互补性强;
- • 不同空间的分子性质分布亦存在差异(Perebyinis & Rognan, 2023),选择合适的空间对项目至关重要。
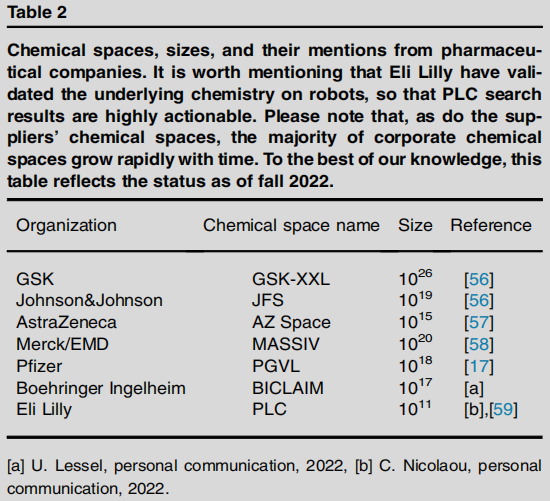
6.2 制药企业专有化学空间(2022 年秋数据)
大型制药公司通常将公有与私有构建块结合,构建远超商业空间规模的专有企业化学空间:
公司 | 化学空间名称 | 规模 |
|---|---|---|
GSK | GSK-XXL | 10²⁶ |
Johnson & Johnson | JFS | 10¹⁹ |
Merck/EMD | MASSIV | 10²⁰ |
Pfizer | PGVL | 10¹⁸ |
Boehringer Ingelheim | BICLAIM | 10¹⁷ |
AstraZeneca | AZ Space | 10¹⁵ |
Eli Lilly | PLC | 10¹¹ |
注:Eli Lilly 已通过机器人验证其底层化学,使 PLC 搜索结果具有极高的可操作性。
七、工业界的实际部署案例
7.1 计算化学空间导航的活性发现
(a)CB2 受体抑制剂(Sadybekov et al., 2022, Nature)
- • 方法:Synthon-based docking,对非枚举的 110 亿化合物空间进行对接;
- • 成果:化合物 505,CB2 抑制活性达亚微摩尔级;
- • 意义:首次证明非枚举超大空间对接的实际可行性。
**(b)ROCK1 激酶抑制剂(Beroza et al., 2022, Nat. Commun.)**
- • 方法:Chemical Space Docking;
- • 成果:抑制剂 22,展示了结构导向化学空间搜索的完整流程。
(c)ROS1 激酶抑制剂(Petrovic et al., AstraZeneca, 2022)
- • 方法:FastROCS 云端筛选 100 亿量级枚举空间;
- • 成果:化合物 31,具有高选择性;
- • 特色:首次在工业规模的云端 GPU 集群上实现百亿级三维形状筛选。
工业界成本效益分析:
- • Merck KGaA 报告称,通过避免暴力枚举方法,计算成本最高节省 90%;
- • 各大公司普遍在初步相似性筛选后引入三维分子叠合或对接进行后处理排序。
7.2 DEL 命中物到临床候选物
化合物 | 靶标 | 研究机构 | 临床状态 |
|---|---|---|---|
GSK'481(GSK2982772) | RIPK1 | GSK | 临床 II 期(炎症性疾病) |
溶解性环氧水解酶抑制剂 | sEH | GSK | 临床 II 期(经分子瘦身优化) |
ENPP2(自分泌素)抑制剂 | ENPP2 | X-Chem | 临床 I 期(特发性肺纤维化) |
更多 DEL 来源化合物正在 2023 年进入临床试验(DICE Therapeutics、Nurix Therapeutics 等)。
八、技术局限性与未来展望
8.1 当前局限的系统梳理
技术 | 核心局限 | 非本质? |
|---|---|---|
按需合成空间 | 环系复杂性不足,立体中心和特殊官能团覆盖有限;对完整 SAR 探索仍力不从心 | ✅ 年增长率高,持续改善 |
生成式 ML | "理论可合成"≠"货架可购买";需要昂贵个性化合成 | ✅ 合成可行性模型持续进步 |
DELs | DNA 兼容反应有限;多样性受约束;固定化靶标需求 | ✅ 反应空间扩展中,非固定化靶标方法成熟中 |
三维搜索 | 精确组合算法难以实现;目前以启发式方法为主 | ⚠️ 理论上更具挑战性 |
8.2 发展趋势预判
近期(1-3 年):
- • 商业按需空间规模持续指数增长,算法跟进;
- • 化学空间对接工具进入工业标配;
- • DEL + ML 的闭环优化流程趋于成熟;
- • 生成式 ML 与合成可行性的一体化设计成为主流。
中期(3-5 年):
- • 量子计算辅助的化学空间搜索(仍处早期探索);
- • 多模态模型整合 2D 结构、3D 形状、蛋白结合、ADMET 等多种信息;
- • DEL 技术向难成药靶标(膜蛋白、转录因子)全面扩展;
- • 设计-合成-测试-分析(DMTA)循环的完全自动化。
九、绿色计算与可持续性思考
这是本综述的一个独特亮点——作者专门从能源效率与可持续性视角审视这些新兴技术:
DEL 的绿色优势:
- • 用料在皮克至纳克量级,远低于传统 HTS 实验室的消耗;
- • 相比大型 HTS 设施,DEL 具有本质性的化学更绿色、废弃物更少的优势。
组合化学空间的绿色计算优势:
- • 以"配方"代替枚举,无需存储 TB 级数据库;
- • 在普通台式机上运行,避免了大型计算集群的能源消耗;
- • 算法的次线性时间复杂度意味着随规模增长,计算成本增速远低于线性。
这一视角与当前学术界和产业界对碳足迹与可持续创新的关注高度契合,为这类技术的长期价值提供了额外论据。
写在最后
10.1 这篇综述的学术定位
本文发表于 Current Opinion in Structural Biology(COSB)的"Drug Discovery New Concepts 2023"专题,由 Andrew R. Leach 和 Alison E. Ondrus 主编。COSB 的综述特点是:篇幅精炼、聚焦前沿、强调作者观点。本文在约 9 页的篇幅内实现了以下目标:
- • 对化学空间生成、搜索与实验技术提供了全景式、有层次的梳理;
- • 附有定量数据支撑(两张重要数据表);
- • 援引了78 篇文献,覆盖面广,重点突出(标注了 7 篇"特别关注"和"最高推荐"文献);
- • 给出了作者对技术局限与未来走向的明确判断。
10.2 需要批判性阅读的方面
- • 作者 Matthias Rarey 与 BioSolveIT GmbH 存在股权关系,Marcus Gastreich 为该公司员工。BioSolveIT 是 SpaceLight、SpaceMACS、Galileo 等工具的开发方,文中对相关工具的介绍需结合利益关系审慎评估;
- • 文章以"综述"定位,对各方法的比较评估较为宏观,缺乏大规模基准测试数据的直接引用;
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-02,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号