J. Am. Chem. Soc. | MolCraftDiffusion:面向计算化学的模块化 3D 分子扩散生成平台

J. Am. Chem. Soc. | MolCraftDiffusion:面向计算化学的模块化 3D 分子扩散生成平台

DrugIntel
发布于 2026-06-29 14:06:40
发布于 2026-06-29 14:06:40

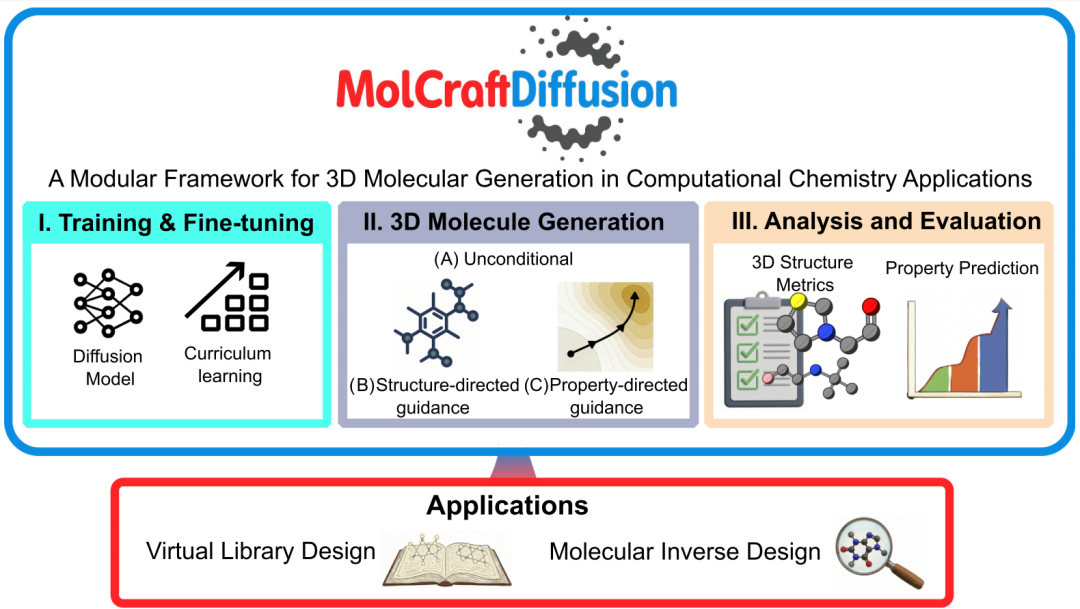
论文信息 标题: Modular Framework for 3D Molecular Generation in Computational Chemistry Applications 作者: Thanapat Worakul, Mohammed Azzouzi, Matthew D. Wodrich, Clémence Corminboeuf* 机构: 洛桑联邦理工学院(EPFL)计算分子设计实验室(LCMD);NCCR-Catalysis 期刊:Journal of the American Chemical Society(JACS) 代码 / 模型 / 教程: GitHub
lcmd-epfl/MolCraftDiffusion· HuggingFacepregH/MolecularDiffusion· 文档站preghosh.github.io/MolCraftDiffusion
核心要点
- • 要解决的问题:3D 分子生成模型理论上比 SMILES / 分子图模型更 物理,但训练成本高、各课题组实现互不兼容,缺乏统一的训练—生成—评测协议。
- • 给出的方案:MolCraftDiffusion——一个分层、解耦、可配置(YAML + CLI)的开源平台,将核心训练逻辑、模型定义、任务实现彼此独立,支持新架构 / 新引导策略 / 新评测指标的"插拔式"集成。
- • 方法学贡献:① 三种课程学习策略(PCL / SPL / HCL)用于扩散模型的高效预训练;② 结构引导(molecular inpainting / outpainting)与性质引导(gradient guidance、classifier-free guidance、混合引导)的统一实现;③ 12 项结构质量指标构成的标准化评测体系。
- • 三个落地案例:不对称环戊二烯基催化剂虚拟库设计、CO₂ 氢化催化剂(IFLP)几何反向设计、单重态裂分发光材料的性质反向设计。
- • 可扩展性验证:在不修改核心代码的前提下,移植了三个架构迥异的外部模型——TABASCO、ADiT、ShEPhERD。
- • 基线模型:E(3)-等变扩散模型 EDM(Hoogeboom et al., 2022),骨干网络为等变图神经网络(EGNN)。
1. 研究背景与动机
1.1 为什么是 3D,而不是 SMILES 或分子图
分子生成模型大致分三条路线:字符串表示(如 SMILES)、图表示、三维坐标表示。前两者发展更成熟,但分子的大多数物理化学性质——催化活性、蛋白-配体结合能、光电性质——本质上由其三维构型决定。因此,能够直接在笛卡尔坐标空间中生成原子位置的模型,理论上提供了一条更"贴近物理"的分子设计路径,对催化剂设计、蛋白-配体结合等任务尤其关键。但 3D 生成的代价是更高的建模复杂度:构象空间庞大、空间约束严格、分子对称性(旋转/平移/置换)必须被显式处理。
1.2 领域内的两个结构性问题
论文开篇明确指出当前 3D 分子生成研究的两个瓶颈:
- 1. 训练成本高:以 EDM 为代表的等变扩散模型在大规模、化学多样的数据集(如 GEOM)上从零训练,收敛困难、计算开销大。
- 2. 生态高度碎片化:自 Hoogeboom 等人 2022 年提出 EDM 以来,后续工作(更鲁棒的架构、结构引导方法、性质引导方法)几乎都是"各自为战"——代码分散在不同仓库,依赖不同的软件环境,使用不一致的评测协议。这直接导致可复现性差、方法间难以公平比较,制约了 3D 生成模型在真实计算化学工作流中的落地。
1.3 已有引导生成方法的局限
在扩散模型基础上,学界已发展出大量"引导生成"技术,大致分两类:
- • 结构引导(scaffold decoration、shape-conditioned generation、lead optimization 等):多数方法将结构约束直接嵌入模型架构,需要专门的训练目标和数据集,难以迁移到其它预训练模型上。
- • 性质引导(梯度引导、无分类器引导等):同样存在类似的"绑定特定模型"问题。
MolCraftDiffusion 的设计目标正是打破这种绑定关系:让引导机制成为可以插在任意(包括第三方)扩散模型上的"插件",而不是某个模型自带的专属功能。
2. MolCraftDiffusion:总体架构
2.1 设计哲学
平台的核心设计原则是分层解耦:核心训练逻辑(trainer/sampler 等底层机制)、模型定义(网络架构)、任务实现(具体的引导策略、评测指标)三者相互独立。新增一种生成架构、一种引导策略或一项评测指标,原则上不需要改动其他层的代码——这也是后文"零代码修改集成 TABASCO / ADiT / ShEPhERD"得以实现的工程基础。
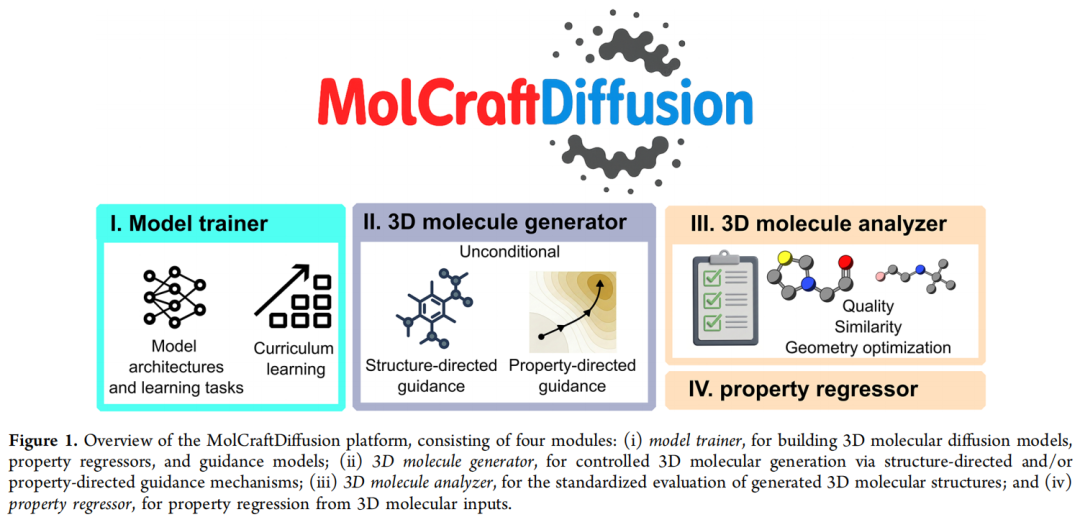
2.2 四大功能模块
模块 | 英文名 | 功能 |
|---|---|---|
(i) 训练器 | Training | 训练扩散模型、性质回归器、引导模型(时间感知的性质回归器);支持从零训练,也支持对预训练模型微调 |
(ii) 生成器 | Generation | 提供结构引导(molecular inpainting / outpainting)与性质引导(梯度引导 / 无分类器引导)两类可组合的受控生成机制 |
(iii) 预测器 | Prediction | 直接从三维分子结构进行性质回归 |
(iv) 分析器 | Analysis | 标准化评测生成结构的化学有效性、几何稳定性、结构完整性与多样性 |
2.3 工程实现细节
- • 交互方式:所有工作流(训练、微调、生成、性质回归、评测)均通过命令行接口(CLI)+ YAML 配置文件驱动,降低了非机器学习背景的化学研究者的使用门槛。
- • 数据层:原生支持多模态分子数据格式——XYZ 坐标文件配合 CSV 元数据、Atomic Simulation Environment(ASE)数据库,以及预处理后的二进制格式;并自动处理图神经网络 / 点云表示所需的 batch collation 逻辑。
- • 化学信息集成:原生对接 RDKit、Morfeus、Cosymlib,可直接引入超越"原子身份"本身的化学相关节点特征(如立体描述符、对称性度量)。
- • 文档与可复现性:每个模块都配有公开教程(
preghosh.github.io/MolCraftDiffusion),论文中每个实验对应的 YAML 配置文件也已存档于 Zenodo,理论上具备逐实验复现的能力。
3. 基础生成模型:E(3)-等变扩散模型(EDM)
MolCraftDiffusion 当前默认使用的生成骨架是 Hoogeboom 等人提出的 EDM(Equivariant Diffusion Model)。
3.1 表示与流程
一个分子被表示为原子类型与三维坐标的组合 x = (h, x)。扩散过程包含两个阶段:
- • 前向过程:逐步向分子的原子属性与坐标添加高斯噪声,直至完全退化为纯噪声;
- • 反向过程:训练一个去噪网络 φ(z_t, t),学习逐步预测并移除每一步添加的噪声,最终从纯噪声重建出合理的分子结构。
去噪网络采用等变图神经网络(EGNN),天然满足三维分子的旋转 / 平移对称性,训练目标是预测噪声与真实噪声之间的均方误差(MSE)。
3.2 采样流程(概述)
- 1. 从标准正态分布采样初始潜变量 z_T;
- 2. 对 t = T, …, 1 逐步执行:用去噪网络计算高斯转移分布的均值与方差,采样得到 z_{t-1};
- 3. 每一步更新后,对坐标分量减去质心(center of gravity),以保持平移等变性;
- 4. 解码最终的 z_0,得到原子类型与坐标 [x, h]。
该流程采用方差保持(variance-preserving)噪声调度来定义信噪比参数 α_t、σ_t 及其逐步比值。完整的扩散方程、网络结构与采样算法细节见原文支持信息。论文同时在 QM9 数据集上对 EDM 架构做了消融研究,考察模型容量、噪声调度方式与原子特征选择的影响(见原文 SI 第 9 节)。
4. 课程学习:让扩散模型训得动、训得快
4.1 动机
扩散模型的去噪目标本质上是高方差的优化问题,直接在 GEOM 这类大规模、化学多样、构象复杂的数据集上从零训练,收敛困难且效果不佳(详见第 5 节的对照数据)。论文借鉴课程学习思想,让模型先掌握简单分子的统计规律,再逐步过渡到复杂结构。
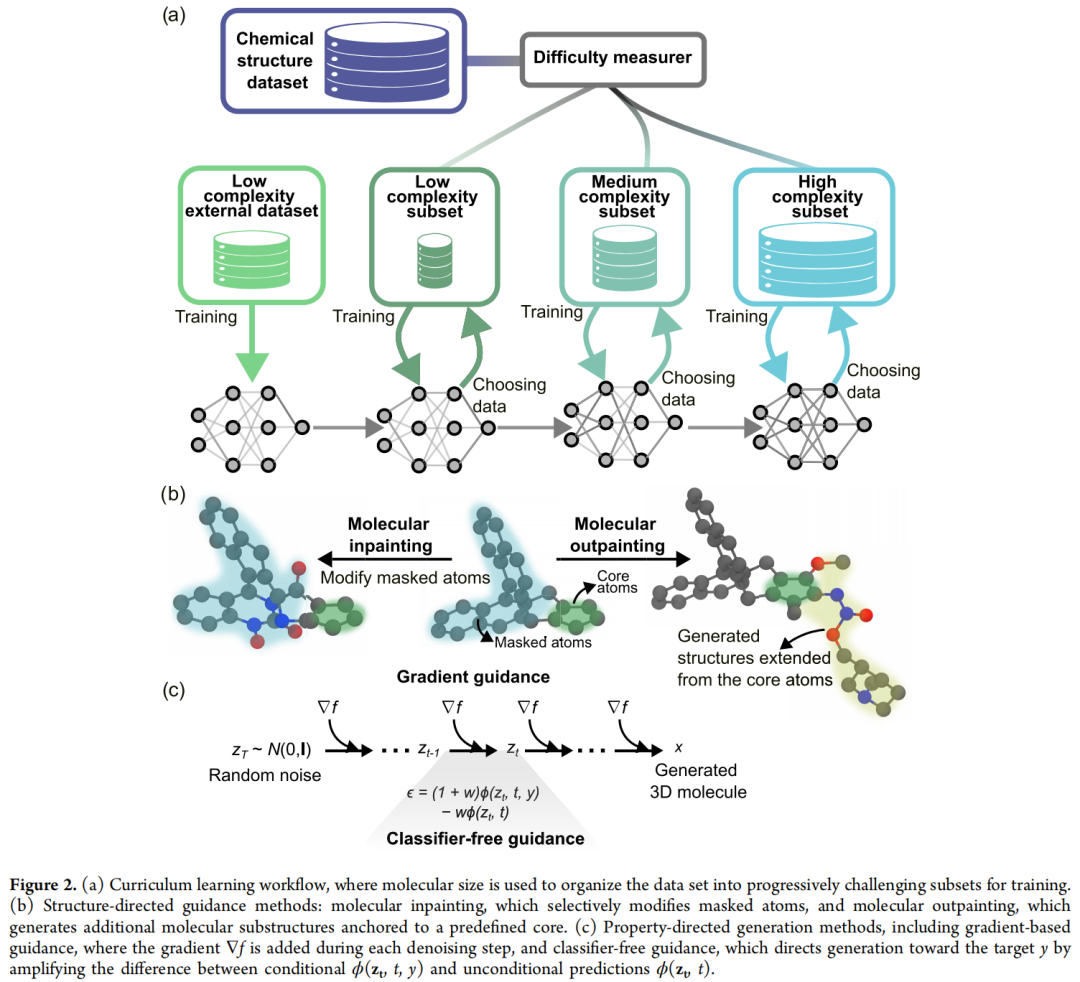
4.2 三种课程学习策略
策略 | 全称 | 机制 |
|---|---|---|
PCL | Predefined Curriculum Learning | 按预先设定的难度标准(分子大小、重原子数、是否含环/支链等拓扑复杂度)对数据集分阶段排序,依固定顺序训练;过程可解释、易实现 |
SPL | Self-Paced Learning | 从在小分子上预训练好的模型出发,每个 batch 内动态丢弃 loss 超过某阈值的样本;阈值随训练逐步放宽,让模型按自身学习节奏推进 |
HCL | Hybrid Curriculum Learning | 先按 PCL 的预定义难度分阶段,每个阶段内部再叠加 SPL 的自适应样本选择——兼顾可解释性与灵活性 |
所有课程学习实验均遵循统一流程:先在 QM9 + VQM24(小分子)组合数据集上预训练 200 个 epoch,再迁移到结构更复杂的 GEOM 数据集,套用上述三种策略之一继续训练。
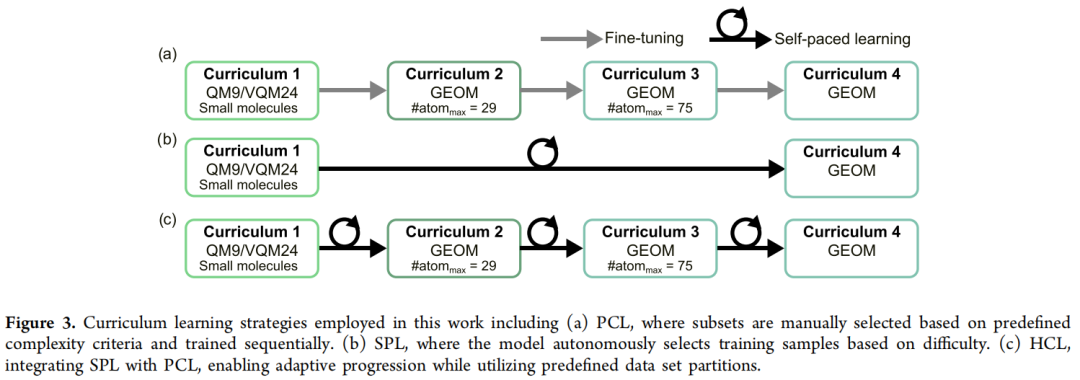
4.3 定量结果(无条件生成 1000 个结构,三次实验均值 ± 标准差)
训练策略 | 化学有效 (%) | 连通 (%) | 有效且连通 (%) | RMSD (Å) | 优化后拓扑完整 (%) | Novelty | Uniqueness |
|---|---|---|---|---|---|---|---|
直接训练 GEOM(无课程) | 9.7 ± 0.1 | 19.3 ± 1.1 | 2.4 ± 0.2 | 1.63 ± 0.04 | 1.1 ± 1.1 | 0.72 | 0.79 |
EDM + PCL | 92.3 ± 0.9 | 76.3 ± 0.8 | 71.9 ± 0.07 | 1.08 ± 0.02 | 83.6 ± 1.5 | 0.68 | 0.78 |
EDM + SPL | 95.7 ± 0.9 | 79.3 ± 0.7 | 77.4 ± 1.2 | 1.00 ± 0.02 | 86.1 ± 1.0 | 0.66 | 0.76 |
EDM + HCL(最优) | 94.8 ± 1.2 | 82.6 ± 1.5 | 79.0 ± 2.1 | 0.97 ± 0.02 | 88.1 ± 2.0 | 0.65 | 0.76 |
EDM + HCL(在多源汇编数据库上继续微调) | 83.4 ± 0.4 | 82.2 ± 1.9 | 73.8 ± 1.6 | 1.03 ± 0.03 | 83.8 ± 1.4 | 0.64* | 0.77* |
*原文正文另给出该行 uniqueness=0.65、novelty=0.77 的描述,与表内列序存在轻微差异,此处两套数值并列标注以供核对。
关键观察:
- • 不加任何课程学习策略,直接在 GEOM 上训练,"有效且连通"比例仅 2.4%,几何优化后保持原拓扑结构的比例仅 1.1%——基本不可用;引入任意一种课程学习策略后,该指标跃升至 70%+ 量级,是数量级的提升,而非边际改善。
- • 三种策略中 HCL 综合表现最佳:有效连通率 79.0%、RMSD 降至 0.97 Å、拓扑保留率 88.1%。
- • trade-off 值得注意:SPL 类策略(SPL、HCL)虽然提升了有效性与几何稳定性,但 uniqueness / novelty 略低于纯 PCL——原因是 SPL 机制会主动丢弃高 loss(即模型"学不会")的训练样本,这在提升收敛质量的同时,客观上压缩了模型实际学到的分子种类范围。这是一个典型的"稳定性 vs. 多样性"权衡,作者也坦诚地指出了这一点,而非只展示对自己最有利的指标。
- • 在 HCL 预训练模型基础上,进一步用 SPL 在多源汇编的 3D 分子数据库上微调,得到了论文实际用于下游应用的预训练基础模型:有效连通率 73.8%,结构多样性(uniqueness 0.65 / 0.77,novelty 0.77 / 0.64,按文中表述)良好——这正是后续三个应用案例所使用的起点模型。
5. 结构引导生成:Molecular Inpainting 与 Outpainting
5.1 设计思路:把图像生成的经典技巧搬到三维分子上
Inpainting(局部重绘)与 outpainting(画布扩展)最初是图像修复 / 补全任务中的技术。论文将这两个概念以"插件式"(plug-and-play)的方式移植到分子扩散模型——不需要为结构约束专门设计模型架构或训练目标,可直接套用在已训练好的无条件扩散模型上。
5.2 Molecular Inpainting(分子局部重绘)
- • 机制:对参考分子的原子属性与坐标施加指定强度 d(denoising strength)的噪声,从而部分擦除结构信息;随后从噪声水平 d 开始重新执行去噪过程,生成新的三维分子。
- • 可控粒度:实现上支持仅对一个"掩码原子子集"加噪,其余原子在整个去噪过程中保持固定——这使得可以做精确的局部编辑(如片段替换、侧链修饰),而不影响分子的其余部分。
- • 关键调节参数 d:d 越大,生成结构与参考结构的差异越大(探索性更强,但失败率上升);d 越小,越倾向于保留原始结构信息(生成结果与参考更相似)。
5.3 Molecular Outpainting(分子骨架扩展)
- • 机制:在扩散的最终时间步 T,将固定的核心骨架与随机初始化的"新增原子"拼接,构成初始潜变量;整个去噪过程中,核心部分的坐标与原子类型保持固定,只对新增区域进行去噪生成。
- • 空间约束:为避免立体冲突,框架额外施加空间约束,确保新生成区域的原子不与核心骨架发生重叠。
- • 两项改进策略(用于提升纯粹"拿来即用"式 outpainting 的生成质量):
- 1. 后期松弛(late-stage relaxation):在去噪过程的最后几步,允许核心部分的原子坐标重新参与去噪(但原子类型仍保持固定),使核心结构能够在空间上微调,与新生成区域更好地融合;
- 2. 专属条件训练目标(Algorithm 4):在前向加噪过程中冻结核心原子的位置和类型,模型被训练为只预测非核心原子上的噪声,从而显式地针对"围绕固定核心补全分子"这一任务进行适配。该训练目标可以直接对已有的无条件预训练模型进行微调,而非从零训练——这一点在第 7.2 节的 IFLP 案例中被证明是决定性的。
5.4 二者的应用场景对比
Inpainting | Outpainting | |
|---|---|---|
典型用途 | 片段替换、侧链 / 局部基团修饰 | 骨架修饰、虚拟化合物库构建、目标几何特征导向的反向设计 |
控制对象 | 指定的掩码原子子集 | 固定的核心骨架 |
关键参数 | 噪声强度 d(保真度 vs. 多样性) | 核心-生成区域的空间约束、(可选)专属微调目标 |
6. 性质引导生成:Gradient Guidance、CFG 与混合引导
在结构约束之外,许多任务(催化活性、激发态能级)需要生成结果在数值上满足目标性质 y。论文实现并系统比较了三种机制:
6.1 梯度引导(Gradient Guidance, GG / classifier-based guidance)
- • 在每一步去噪时,使用目标函数 f 相对于当前潜变量 z_t 的梯度 ∇f(z_t) 来扰动采样轨迹,引导生成方向逼近目标性质。
- • 该方法本身不需要微调扩散模型,但要求存在一个可微、且作用于与扩散模型一致的分子表示(笛卡尔坐标 + 节点属性)的目标函数。为此,论文额外训练一个共享 EGNN 骨干的"引导模型" f_θ(z_t, t),通过监督回归——在干净结构 x₀ 上按扩散噪声调度施加扰动得到 z_t,再让模型学习预测对应的真实性质 y——来获得这个可微目标函数。
- • 特点:该引导模型与具体扩散模型的噪声调度、参数化方式绑定,更换扩散架构需要重新训练;引导强度与梯度裁剪阈值需要手动权衡,避免生成轨迹偏离训练分布过远。
6.2 无分类器引导(Classifier-Free Guidance, CFG)
• 不依赖外部分类器/回归器,而是直接对扩散模型本身做适配:扩展 EGNN 编码层的输入维度以容纳条件变量 y(额外通道随机初始化),并在训练时以一定概率(通常 0.1–0.2)将条件值替换为预定义的"空值 token",从而让同一模型既能无条件生成、也能按条件生成。
• 推理阶段的核心公式:
ε̃ = (1 + w) · φ(z_t, t, y) − w · φ(z_t, t)其中 φ(z_t, t, y) 是条件预测、φ(z_t, t) 是无条件预测,w ≥ 0 为引导强度(guidance scale)。w 越大,生成结果越偏向满足条件 y,但同时越可能牺牲结构有效性。
• CFG 同样支持多目标条件组合(多性质同时引导),细节见原文 SI。
6.3 混合引导(CFG / GG)
- • 将条件扩散模型(CFG)与梯度引导(GG)并联使用:条件模型先把生成方向"扳"到大致正确的区域,梯度引导再做精细的轨迹微调。需要分别设定 CFG 与 GG 各自的引导强度,以及梯度引导在哪些(或多大比例的)去噪步骤上生效。
- • 在第 7.3 节的单重态裂分案例中,混合引导在"性质命中率"与"结构有效性"两个维度上都取得了三者中最好的综合表现。
7. 应用案例深度解析
论文用三个真实计算化学问题验证框架的实用性,难度递进:从相对成熟的虚拟库构建,到几何反推,再到需要量子化学验证的激发态性质反推。
7.1 案例一:不对称环戊二烯基(Cp)配体虚拟库设计
化学背景:环戊二烯基(Cp)配体与 Co/Rh/Ir 等第 9 族金属络合后,可催化高对映选择性的二氢异喹啉酮类反应等不对称 C–H 官能化反应。但这类配体合成路线复杂,实验上很少进行系统性的大规模筛选——这正是虚拟库设计能够填补的空白:构建一个允许系统分析"立体效应—活性/选择性"关系的数据库。
立体化学模型:以 1,1′-联-2-萘酚环戊二烯基(BINOL-Cp) 为起始骨架,其不对称诱导原理依赖三个结构元素——"后墙""侧墙""天花板",分别由 BINOL 部分(后墙、侧墙)与 Cp 环上 R¹/R²/R³ 位点的取代(天花板)构成。论文采用以金属中心为球心、半径 5 Å 的象限 / 八分体(quadrant/octant)埋藏体积分析来量化立体环境,其中 Q1、Q4、O1、O4 等区域对应底物接近金属中心的关键方向。
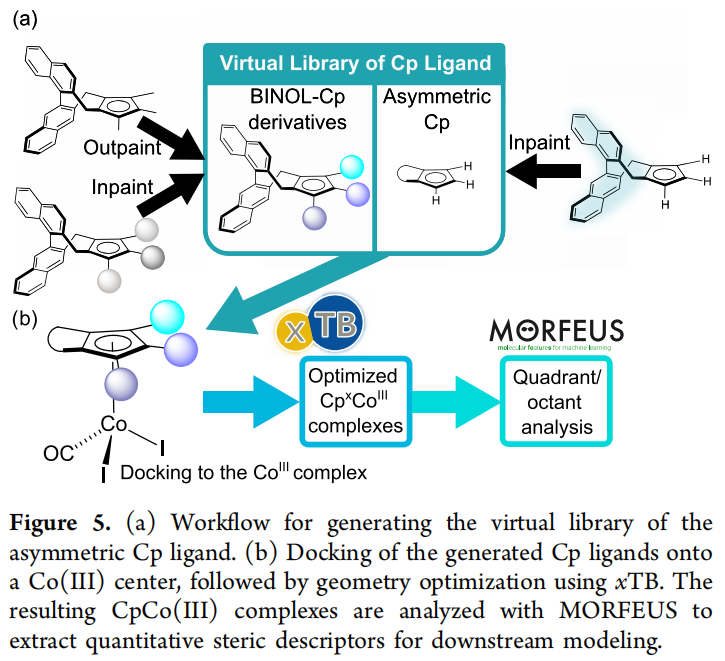
生成流程与结果:
- 1. 从裸 BINOL-Cp 骨架出发,使用 outpainting 在 R¹、R²、R³ 位点生成取代基:初始生成 2,500 个结构,其中 745 个通过"满足价键规则 + 总电荷为 -1"的化学有效性检查(成功率 29.8%)。
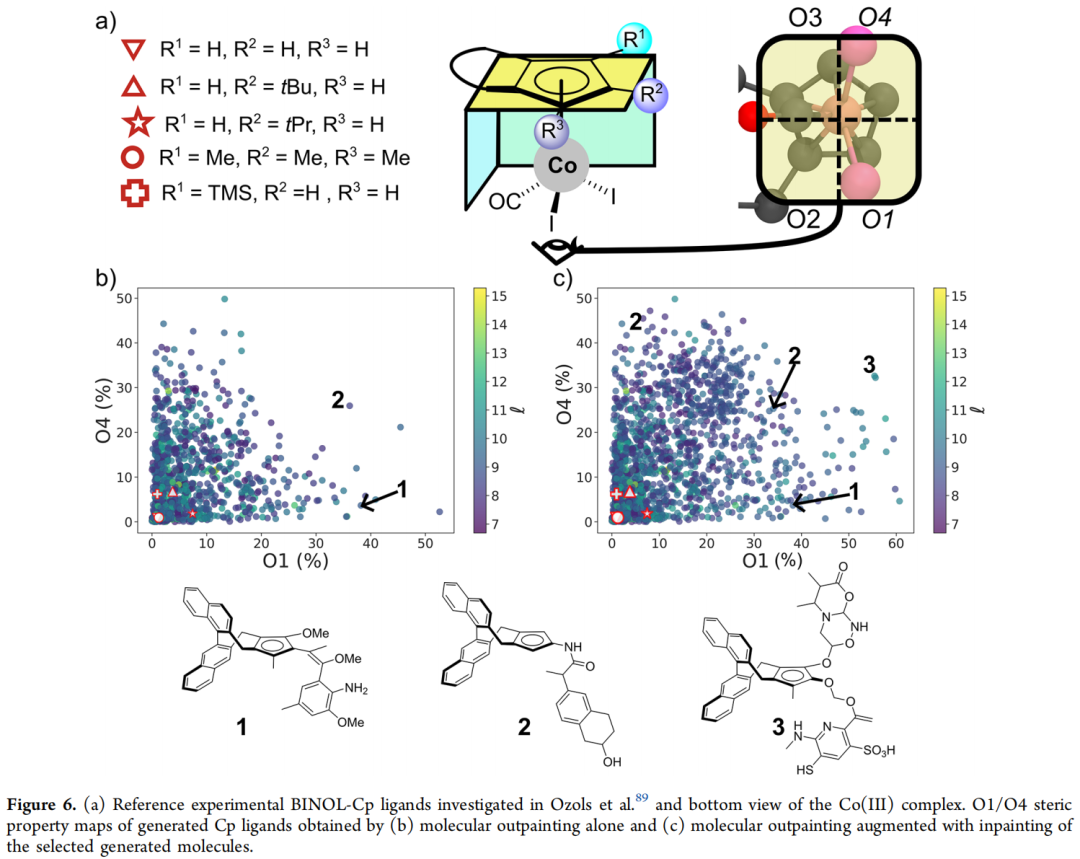
- 2. 将有效配体对接到 Co(III) 络合物上,经 GFN2-xTB 几何优化后,用 Morfeus 计算立体描述符。结果显示:AI 生成的 BINOL-Cp 衍生物在 O1/O4 立体性质图谱上覆盖了远比现有实验配体更宽广的区域;而真实合成出来的配体几乎全部聚集在"低立体阻碍"的一角。
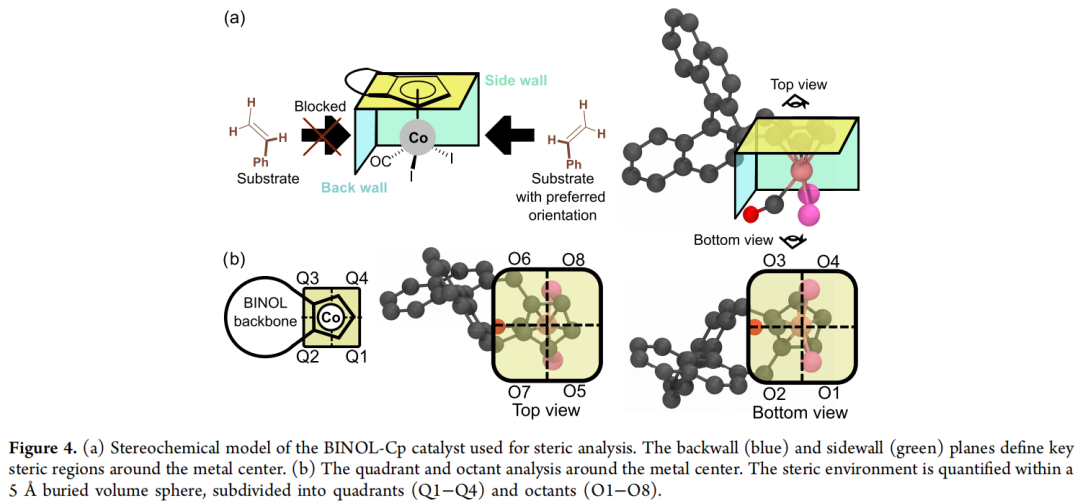
- 3. 针对初始生成结果仍稀疏覆盖的区域(O1/O4 图谱右上角),论文选取两个邻近空白区的种子结构(标注为配体 1 与 2),改用 inpainting 仅对 R¹/R²/R³ 做局部变动、骨架保持不变:在 1,000 个新生成结构中,额外获得 366 个有效结构,成功填补了此前稀疏的区域。生成的代表性配体(如配体 3)可以从几乎所有方向屏蔽金属中心,呈现出极端的立体拥挤构型。
- 4. 为探索"后墙/侧墙"(Q2/Q3 区域)的多样性,论文进一步对 BINOL 骨架本身(而非仅取代基)施加 inpainting,保持 Cp 环与氢取代基固定,并限制生成分子的尺寸与原骨架相近。该过程的核心调节参数仍是去噪强度 d:当 d = 0.3 时,有效配体比例最高(87%),但多数与原始 BINOL-Cp 结构高度相似,结构变化有限;随着 d 增大,生成的骨架变得更多样(包括与 BINOL 显著不同的结构),但有效率相应下降(详见原文表 S4)。代表性结构(配体 4、5、6)展示了从"Q3 拥挤、Q2 开放"(含吲哚基团)到"Q2、Q3 同时拥挤"等多种立体组合。
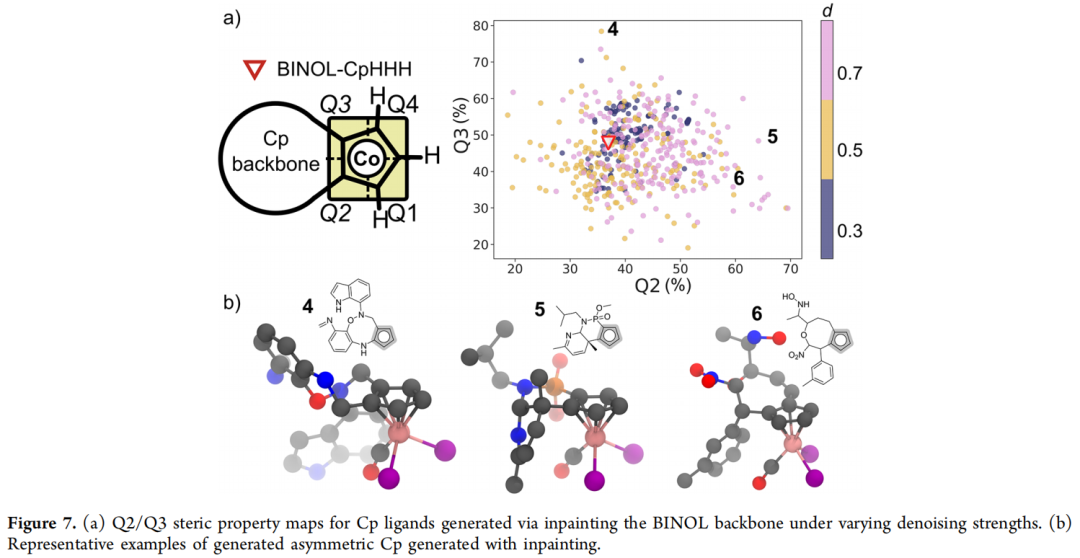
小结:本案例较为成功地展示了"outpainting 广覆盖 + inpainting 精补漏"这一组合策略在虚拟库构建任务上的有效性,是论文中执行最顺利的应用场景。
7.2 案例二:CO₂ 氢化催化剂——分子内受阻路易斯酸碱对(IFLP)的几何反向设计
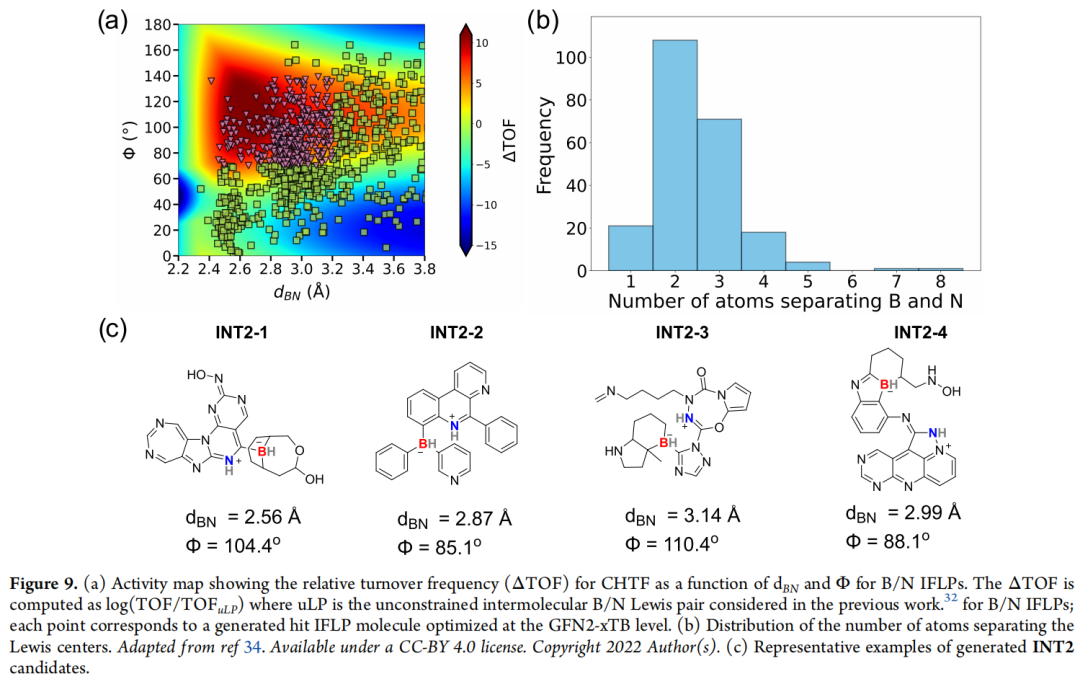
化学背景:受阻路易斯酸碱对(FLP)是路易斯酸与路易斯碱在同一分子框架内被空间因素阻止直接配对中和、从而保有反应活性的体系,可用于活化 H₂、CO₂ 等小分子。在 CO₂ 催化氢化制甲酸盐(CHTF)的反应循环中:IFLP 先活化 H₂ 生成关键中间体 INT2(路易斯酸结合 H⁻、路易斯碱结合 H⁺),随后与 CO₂ 结合生成 INT3,最终形成甲酸盐产物 INT4 并使催化剂再生。前人研究(Dotson et al., 2023;Das et al., 2022/2024)已确立 INT2 的两个几何参数——B–N 距离 d_BN 与 B–H/N–H 键夹角 Φ——与催化活性(以相对转化频率 ΔTOF 衡量)高度相关。
设计目标:将生成的 INT2 中间体限定在高活性区间 d_BN = 2.4–3.2 Å、Φ = 70–140°。有效性判定遵循三条标准:① 所有原子满足标准价键规则;② 核心骨架中的氢化物 / 质子分别且只与 B / N 中心成键;③ B 与 N 中心属于同一连通分子。
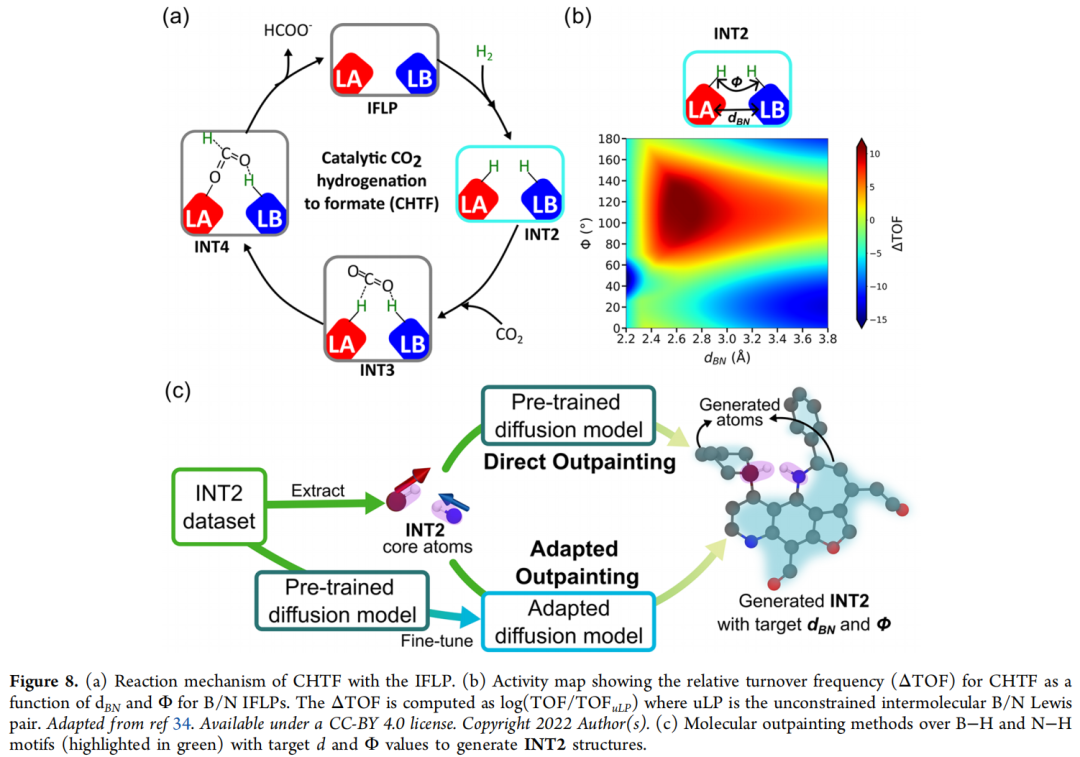
实验过程与关键数字:
方法 | 起始骨架 | 有效 INT2 比例 |
|---|---|---|
直接 outpainting(预训练模型,未微调) | B–H/N–H 核心(最小骨架) | 5.2% |
直接 outpainting + 扩展骨架(纳入 Lewis 中心邻近原子) | 扩展骨架 | 9.8% |
自适应外推(专属训练目标微调 200 epoch) | 目标骨架(d_BN=2.82 Å, Φ=94.8°,源自 CoRE MOF 2019 数据库) | 38.1% |
自适应外推 | 更常见骨架(d_BN=2.67 Å, Φ=33.45°) | 46.7% |
直接 outpainting 表现不佳的原因被归结为:B–H/N–H 这一核心骨架本身空间结构稀疏、缺乏刚性支撑(不同于上一案例中刚性的 Cp 环骨架),导致生成模型容易产生畸变结构。引入"自适应外推"——即针对 outpainting 设计专属训练目标、在微调中冻结核心原子——后,有效率提升了一个数量级。
几何优化后的"双料"评估:用 GFN2-xTB 对生成结构做几何优化后,需同时满足"化学完整性保留"(优化后拓扑未被破坏)与"几何指标命中目标区间"两个条件才算真正的 postoptimization hit:
起始骨架 | 优化后拓扑完整 | 优化后命中目标区间 |
|---|---|---|
常见骨架 | 83.8% | 18.6% |
目标(稀有)骨架 | 64.1% | 25.7% |
这是一组值得专门指出的反直觉结果:尽管常见骨架在优化后结构完整性更高,但其几何指标命中目标区间的比例却更低;而本身在数据集中代表性不足的目标骨架,尽管结构稳定性稍差,命中率反而更高。这提示骨架选择本身就是一个需要权衡的设计变量,而非可以忽略的细节。
论文进一步指出:高活性候选分子中,B、N 中心之间多间隔 2–3 个原子(对应 cis-vicinal 或 ansa 型排布),与既往工作(Das et al., 2024)的结论一致;但生成的 IFLP 候选总体表现出比训练集更高的合成复杂度评分(SCScore、SAScore),提示合成可行性仍是短板;个别结构含有奇异基序(quinoidal 型结构、多氮杂芳环)。此外,作者尝试在分子表示中加入额外原子特征以提升生成质量,结果显示优化前的有效率确有提升,但优化后的有效率 / 命中率反而下降(详见原文表 S4)——这与下文单重态裂分案例中"加特征即提升"的结论形成对照,说明该方法的增益高度依赖具体化学体系,IFLP 这种分子尺寸更大、约束更复杂的体系,改进效果尚不明确。
局限性(作者自陈):当前的引导框架尚未纳入路易斯中心的酸碱性等化学性质约束,原因是相关结构—性质关系的泛化建模本身仍具挑战;几何优化后的"双料命中率"总体偏低,作者认为主动学习(将命中结构迭代加入训练集)是值得探索的改进方向。
7.3 案例三:单重态裂分(Singlet Fission)发光材料的性质反向设计
化学背景:单重态裂分是指一个被吸收的光子裂变为两个三重态激子的物理过程,若能用于光伏器件,理论上可以突破单结太阳能电池的 Shockley-Queisser 效率上限。一个分子要被视为可行的单重态裂分(SF)色基团,必须在几何优化之后仍同时满足三条竖直激发态判据:
- • 近热中性的裂分驱动能:ΔSF ≡ E(S1,ve) − 2·E(T1,ve) ≳ −1 eV
- • 三重态能级落在硅基太阳能电池的吸收响应区间:E(T1,ve) > 1.1 eV
- • 能吸收太阳光谱中丰富的光子:E(S1,ve) < 3.8 eV
这是一个比前两个案例更苛刻的设计任务:生成模型必须同时产出合理的基态几何 和 经量子化学计算验证后仍成立的激发态性质。
数据与代理模型:论文使用 FORMED 数据库(11.7 万个实验报道的有机晶体结构,配有 TD-DFT@ωB97X/6-31G* 计算的基态/激发态性质)训练了一个 EGNN 代理模型,直接从三维结构预测 E(S1,ve) 与 E(T1,ve),同时服务于梯度引导的目标函数计算与候选结构的快速初筛;最终验证则通过完整的 GFN2-xTB 几何优化 + TD-DFT 重新计算完成。
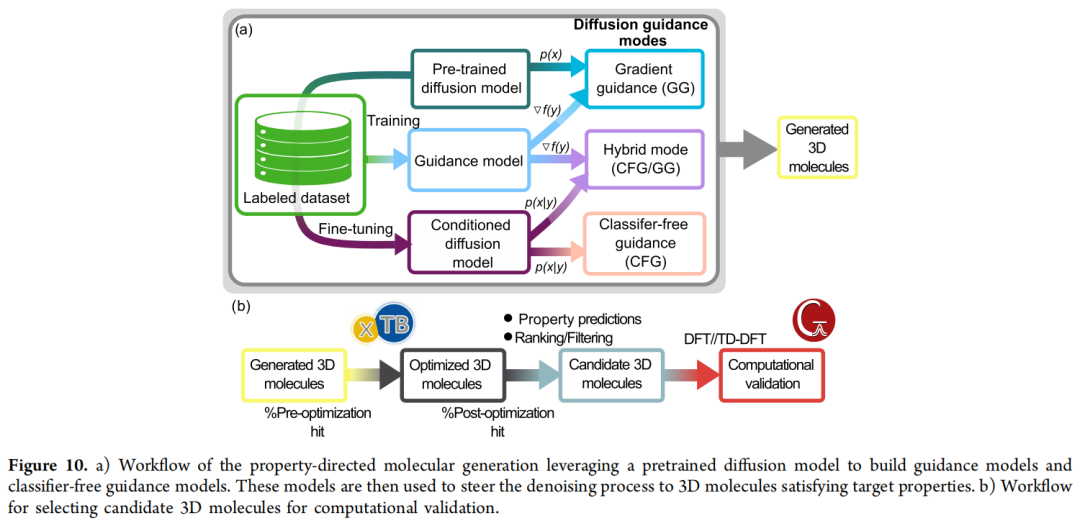
三种引导方式的对照实验(各生成 1,000 个分子):
引导方式 | 化学有效率 | 优化前命中率 | 优化后命中率 | 与优化结构的 RMSD |
|---|---|---|---|---|
梯度引导(GG) | 65.5% | 62.9% | 13.6% | 1.02 Å |
无分类器引导(CFG) | 89.1% | 27.8% | 27.8% | 0.53 Å |
混合引导(CFG/GG) | 84.7% | 80.8% | 43.9% | 0.57 Å |
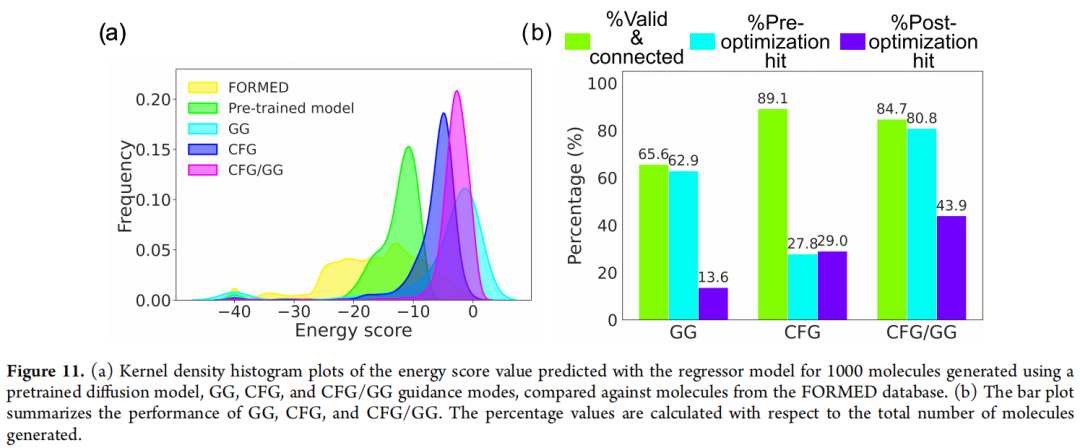
解读:
- • CFG 生成的分子化学有效率最高、几何也最稳定(RMSD 最小),但由于 FORMED 数据库中天然满足 SF 条件的分子样本有限,条件信号偏弱,因此把能量分布"推"向极端区域的能力有限;
- • GG 凭借显式梯度,能在优化前把命中率推得很高(62.9%),但代价是频繁扭曲分子几何(RMSD 高达 1.02 Å),导致绝大多数"优化前命中"在几何优化后失效,最终命中率反而最低;
- • 混合引导结合了两者优势——CFG 提供大方向上正确、结构合理的起点,GG 再做精细的性质微调——在优化前命中率(80.8%)与优化后命中率(43.9%)两个维度上都是三者中表现最好的方案,且结构精度(RMSD 0.57 Å)与 CFG 接近。
多样性与新颖度:三种引导方式生成结构的 uniqueness 介于 0.70–0.80、novelty 介于 0.65–0.70,明显高于 FORMED 数据库中已知满足 SF 条件的分子子集本身的 uniqueness(仅 0.49)。论文特别将其与此前团队基于强化学习(REINVENT 系列方法)做同类设计的结果进行对比:强化学习方法虽然命中率也不低,但生成结果容易在单次实验内集中于很窄的化学空间,往往需要额外的结构约束和多次独立运行才能保证探索的广度;相比之下,扩散模型 + 混合引导这一路线天然产出了更分散、更具探索性的候选集合。
最终筛选与验证:以混合引导生成结果中、按优化后 SF 评分排序的前 150 个分子为分析对象:134 个满足竖直激发态判据;进一步采用更严格的绝热激发态标准(S1,ad − 2T1,ad ≥ 0 eV),最终确认 69 个为可靠的单重态裂分候选(经 TD-DFT@ωB97X-D/6-31G(d) 验证)。结构上呈现出富勒烯型(fulvenoid)、偶氮氧(azoxy)等异质环基序,与既往研究的结论一致。
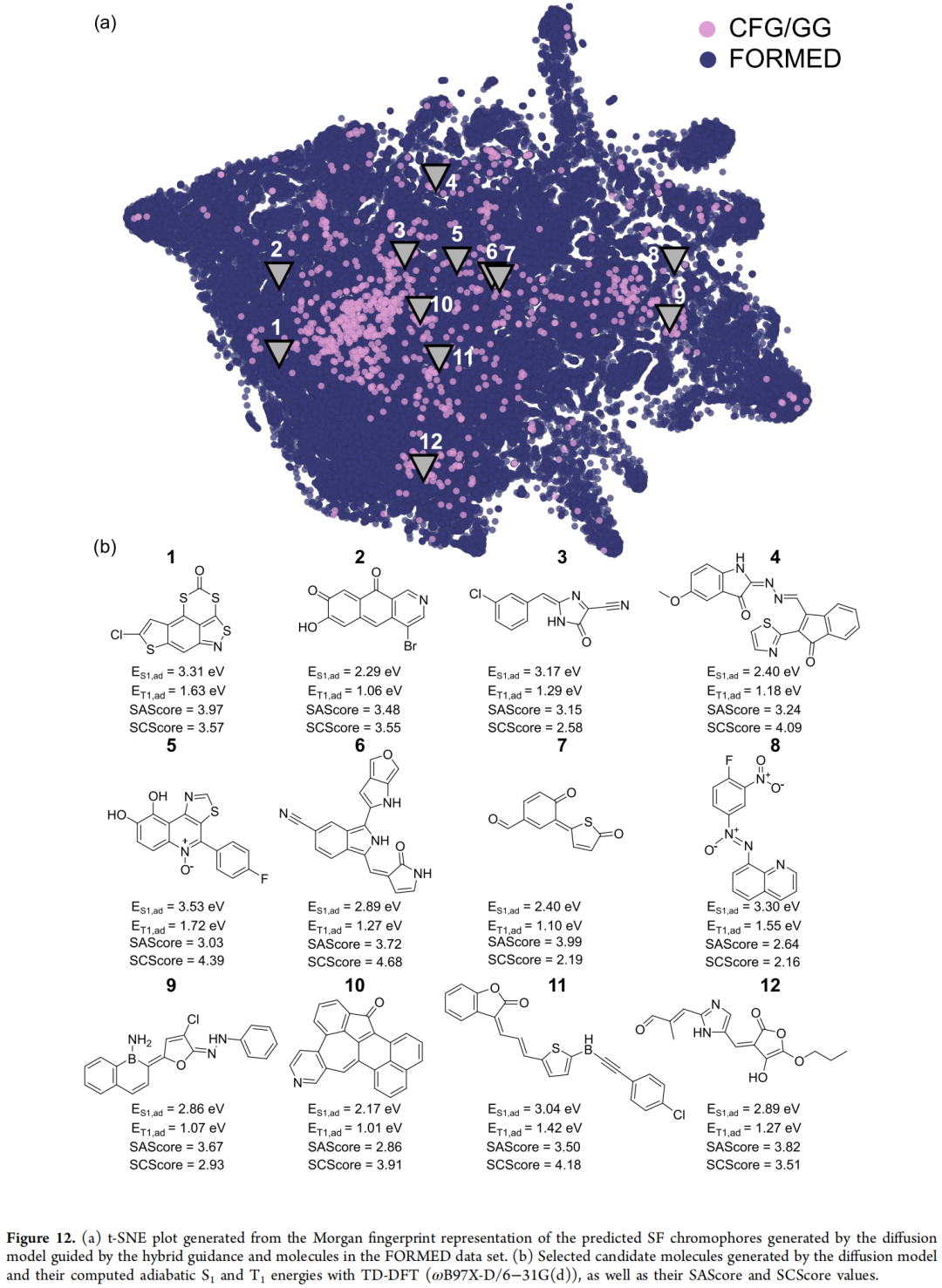
作者自陈的局限:部分高分候选存在"好看但不真"的问题——例如偶氮开关基团中出现反向 π 共轭、自由硼中心易发生水解、部分异质环在合成上极不现实——而这些缺陷恰恰不被 SAScore、SCScore 这类标准合成可行性评分捕捉,暴露出现有合成性评分体系在分子稳定性判断上的盲区。
进一步改进尝试:在分子表示中引入额外原子特征,并将 SCScore 作为额外的条件变量与目标能级一起引导生成,结果使生成分子的平均 SCScore 从 3.9 降至约 3.4,结构上也观察到不再出现三周期元素,不稳定/反应性官能团(如烯酮、羟基肼、Se–S 基序)明显减少——但该改进伴随轻微的命中率下降,体现出"性质优化"与"合成可行性约束"之间的典型权衡。与 7.2 节中 IFLP 体系的类似尝试相比,这一改进在 SF 体系中效果更明确,论文也据此提示:附加原子特征 / 条件变量的增益因化学体系而异。
7.4 三个案例的横向对比
案例一:Cp 配体虚拟库 | 案例二:IFLP 几何反推 | 案例三:单重态裂分 | |
|---|---|---|---|
引导类型 | 结构引导(outpainting + inpainting) | 结构引导(outpainting + 自适应微调) | 性质引导(GG / CFG / 混合) |
设计目标 | 拓展立体多样性覆盖 | 命中特定几何参数区间 | 命中多条激发态能级判据 |
是否需微调预训练模型 | 否(直接可用) | 是(直接使用效果不佳) | 是(CFG/混合需微调) |
验证手段 | xTB 优化 + Morfeus 立体描述符 | GFN2-xTB 优化 + 几何参数复测 | GFN2-xTB 优化 + TD-DFT 验证 |
核心结论 | 框架"拿来即用"已能产出有价值结果 | 稀疏/柔性核心骨架对直接 outpainting 是挑战,需专属微调 | 混合引导综合最优,但合成可行性评估仍有盲区 |
8. 标准化结构质量评测体系
论文构建了一套覆盖化学有效性、几何合理性、稳定性与多样性的 12 项指标评测体系,作为框架"分析器"模块的核心功能:
基础有效性检查
- 1. 化学有效性(Chemical validity, %)——是否满足标准价键规则
- 2. 连通性(Connected, %)——是否构成单一连通图
- 3. 有效且连通(Chemically valid and connected, %)——同时满足以上两者
PoseBusters 几何合理性检查(与参考几何值对比) 4. 键长 / 键角(Bond lengths/angles) 5. 环平面性(Ring flat) 6. 双键平面性(Double bond flatness) 7. 无立体冲突(No steric clash) 8. 内能合理性(Internal energy)
几何优化前后的一致性 9. 平均 RMSD(Å)——生成结构与 GFN2-xTB 优化结构之间的均方根偏差 10. 拓扑完整性保留(Intact chemical topology, %)——优化后是否仍保持原拓扑
化学空间多样性(基于 Morgan 分子指纹的 Tanimoto 相似度) 11. Uniqueness——生成批次内部的相互差异性 12. Novelty——相对训练数据集的新颖程度
这一体系的价值在于:所有架构(无论是框架自带的 EDM,还是后续移植的 TABASCO / ADiT / ShEPhERD)共享同一套评测协议,使得不同模型之间的横向比较第一次具备了可信的统计基础——这正是论文反复强调的"标准化平台"价值的具体落地。
9. 平台可扩展性验证:移植三个外部模型
为验证"分层解耦"架构的真实可扩展性,论文在不修改核心代码的前提下,将三个架构迥异的文献模型接入 MolCraftDiffusion:
模型 | 架构类型 | 训练方式 | 集成定位 |
|---|---|---|---|
TABASCO(Vonessen et al., 2025) | 基于 Transformer、采用 flow-matching 框架 | GEOM 数据集训练 600,000 步 | 通用无条件生成器,与 EDM-HCL 横向对比 |
ADiT(Joshi et al., 2025) | All-atom 隐空间扩散 Transformer | GEOM 数据集训练 600,000 步 | 通用无条件生成器,与 EDM-HCL 横向对比 |
ShEPhERD(Adams et al., ICLR 2025) | SE(3)-等变扩散模型,联合建模 3D 结构与形状 / 静电势曲面 / 药效团等交互特征 | 加载原始预训练权重 ShEPhERD-GDB17 | 验证专精任务(药效团条件生成 / 生物等排体设计)的可移植性 |
TABASCO 与 ADiT 在与 EDM-HCL 相同的训练 / 生成 / 评测协议下进行评估,结果(原文表 S3)显示其性能与 EDM-HCL 以及文献中的对照模型(Nikitin et al., 2025)相当,证明了平台在通用无条件生成任务上的横向比较能力。
ShEPhERD 的集成更进一步——论文同时将其专属的评分函数(基于交互特征的一致性度量)也整合进分析模块,并通过加载原始预训练权重验证自一致性:生成结构的形状与静电势曲面与直接计算得到的真实曲面高度吻合,与原作者报告的结果一致;药效团的自相似度相对形状/ESP偏低,但仍显著高于随机分子基线。
这一节是全文中最直接支撑"模块化架构具备真实可扩展性"这一核心论点的实证部分。
10. 综合讨论与局限性
整理全文中作者自陈以及可推断的方法学局限,供读者批判性参考:
- • 化学约束的覆盖仍不完整:当前引导框架未将路易斯中心酸碱性等需要复杂结构—性质关系建模的化学性质纳入引导目标,原因是相关泛化建模本身存在困难(IFLP 案例)。
- • 合成可行性评分存在盲区:SAScore、SCScore 等标准评分无法捕捉某些"看起来正常但实际不稳定/不现实"的结构缺陷(如易水解的硼中心、反向共轭的偶氮基团),意味着仅靠这些指标筛选候选分子并不充分。
- • 效果的体系依赖性:附加原子特征、引入合成性评分作为条件变量等改进策略,在单重态裂分案例中效果明确,但在 IFLP 案例中结论并不一致——提示方法的增益高度依赖具体化学体系的分子尺寸与约束复杂度,而非"放之四海而皆准"。
- • 稀疏/柔性核心骨架对 outpainting 仍是挑战:相比刚性的环戊二烯骨架,IFLP 中稀疏的 B–H/N–H 核心对"拿来即用"式 outpainting 的挑战明显更大,必须依赖专属微调才能获得可用的命中率,这是当前框架在通用性上的一个边界。
- • 几何优化后的"双料命中率"总体偏低:即便是综合表现最好的混合引导方法,单重态裂分案例下的优化后命中率也只有 43.9%;IFLP 案例的最佳双料命中率约 25.7%。作者认为主动学习(将命中结构迭代纳入训练)是值得探索的提升路径,但尚未在本文中实证。
这些局限性的坦诚披露,本身是这篇方法学论文质量的一个体现——它没有把三个案例包装成"完美闭环",而是把每一处不足都摆在了桌面上。
11. 总结
MolCraftDiffusion 的核心贡献并不在于提出了一个性能更强的新生成模型,而在于为 3D 分子扩散生成研究提供了一套可复用、可比较、可扩展的工程基础设施:
- • 通过课程学习,把"扩散模型在化学多样数据集上训不动"这一痛点,转化为一个有明确方法论支撑、效果可量化(2.4% → 79.0%)的工程问题;
- • 通过结构引导与性质引导的统一实现,把原本分散在各篇论文里、彼此不兼容的"控制生成"技巧,整合进同一套可插拔的接口;
- • 通过三个真实计算化学案例的端到端验证(生成 → 计算化学验证),展示了从"分子长得像不像"这种表层评测,走向"分子是否真的满足下游应用判据"的可能路径;
- • 通过零代码修改移植三个外部模型,对"模块化、可扩展"这一架构主张给出了具体可检验的证据。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号