J. Chem. Theory Comput. | 预训练机器学习势能在分子模拟中的准确性与计算效率基准评测

J. Chem. Theory Comput. | 预训练机器学习势能在分子模拟中的准确性与计算效率基准评测

DrugAI
发布于 2026-06-24 13:22:20
发布于 2026-06-24 13:22:20
近年来,大规模量子化学数据库的出现推动了预训练机器学习原子间势能(Machine Learning Interatomic Potentials,MLIPs)的快速发展。越来越多的基础模型能够覆盖广泛化学空间,并支持复杂分子体系模拟。然而,随着模型数量迅速增加,研究人员面临一个新的问题:究竟应该选择哪一种势能模型用于具体分子模拟任务。
研究人员对15种主流预训练MLIP进行了统一基准测试,从预测精度、计算速度、显存消耗以及分子动力学稳定性四个方面进行系统评估。结果表明,模型参数规模和训练数据规模与预测精度呈现显著相关关系,而显式引入库仑长程相互作用项并未表现出明确优势。同时,不同模型在速度和显存需求方面存在巨大差异,其影响因素不仅来自模型规模,也与模型架构密切相关。研究建立了当前分子模拟领域最全面的预训练MLIP基准体系,为研究人员选择合适势能模型提供了客观依据,也揭示了影响模型性能的关键因素。

传统分子动力学模拟通常依赖经验力场或量子化学计算。经验力场计算速度快,但难以达到高精度量子化学水平;而密度泛函理论等量子化学方法虽然准确,却往往计算成本极高,难以应用于长时间尺度和大体系模拟。
机器学习势能的发展为解决这一矛盾提供了新思路。通过利用大规模量子化学数据训练神经网络模型,可以在接近量子化学精度的同时获得接近经典力场的计算效率。近年来,随着SPICE、OMol25等大型数据集的发布,一批被称为“基础势能模型(Foundation Potentials)”的预训练MLIP相继出现。
然而,不同模型通常采用不同测试集、不同评价指标以及不同硬件环境进行性能展示,导致研究人员难以进行客观比较。此外,大多数公开评测主要关注预测精度,而忽略了计算速度、显存占用以及模拟稳定性等实际应用中同样重要的指标。
研究人员因此开展本研究,希望通过统一测试框架对当前主流预训练MLIP进行系统评估,为未来模型开发和实际应用提供参考。
方法
研究纳入15种具有代表性的预训练机器学习势能模型,包括AceFF、AIMNet2、Egret-1、FeNNix-Bio1、MACE系列、Orb-v3以及UMA系列等。所有模型均支持广泛元素覆盖,并能够进行能量守恒的分子动力学模拟。研究采用SPICE测试集评估预测精度,该测试集包含800个未参与训练的小分子、大分子、五肽以及蛋白质-配体二聚体构象。随后利用统一硬件平台(NVIDIA H100 GPU)测量不同体系规模下的模拟速度和显存占用情况。同时,研究通过100 ps高温分子动力学模拟评估各模型的数值稳定性和化学稳定性,从而实现对准确性、效率和可靠性的综合评价。
结果
构建统一的预训练MLIP评测体系
研究首先建立了一套统一评测框架,用于比较当前主流机器学习势能模型。
与以往工作不同,本研究不仅评估预测误差,还同时考虑计算效率、显存需求和模拟稳定性。测试集覆盖小分子、大分子、多肽以及蛋白质相关复合体系,同时包含中性分子和带电分子,从而能够更全面反映模型在真实应用中的表现。
研究指出,以往广泛使用的MD17等基准主要包含20个左右原子的分子,而现实药物分子、生物大分子片段和蛋白质相互作用体系通常远比这些测试对象复杂。因此,更接近真实应用场景的评测对于基础势能模型的发展具有重要意义。
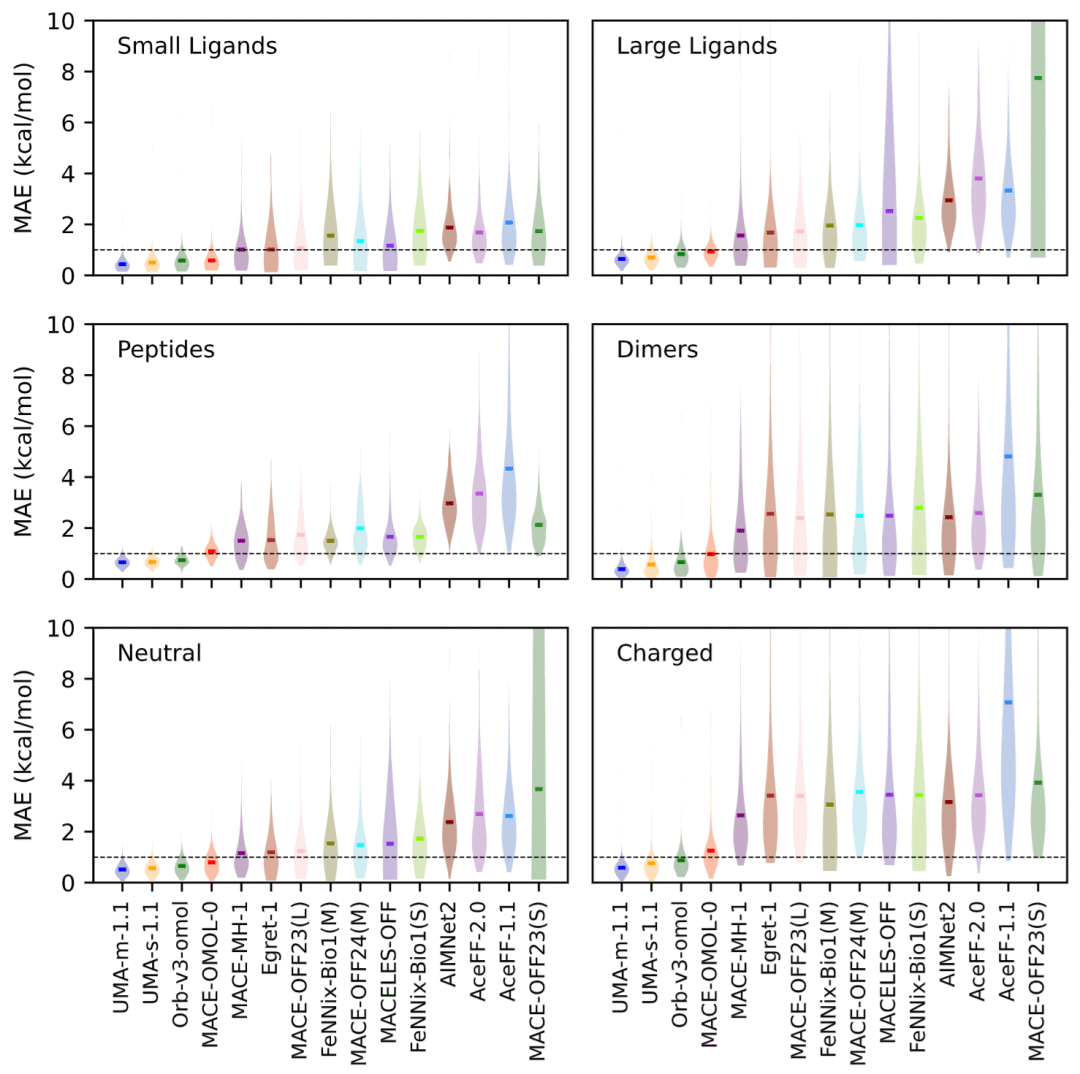
图1: 不同预训练机器学习势能模型在SPICE测试集上的总体误差分布。
UMA与Orb模型取得最高预测精度
在准确性评估中,研究利用构象间能量差作为主要评价指标,以减少不同量子化学理论水平带来的影响。
结果显示,UMA-m-1.1获得全体模型中的最佳表现,其整体平均绝对误差约为0.53 kcal/mol。紧随其后的是UMA-s-1.1和Orb-v3-omol,两者误差均保持在化学精度标准以内。
研究进一步发现,UMA系列和Orb系列不仅整体误差较低,而且在不同类型体系之间表现较为均衡。相比之下,一些模型虽然在小分子上表现优异,但在大分子或多肽体系中的误差显著增加。特别值得注意的是,MACE-OFF23(S)虽然在小分子上具有较高精度,但当分子规模增加时误差迅速扩大,显示出较差的尺度泛化能力。
研究认为,优秀模型不仅需要在平均误差上表现突出,更需要在不同化学空间中保持稳定性能。
模型规模与训练数据规模决定预测精度
研究进一步分析了影响预测精度的关键因素。结果显示,模型参数数量与预测误差之间存在明显相关关系。总体趋势表明,参数规模越大,预测误差越低。同时,训练数据集规模也与模型性能密切相关。例如,UMA系列模型拥有最大的参数规模和训练数据规模,因此获得最高精度。而训练于OMol25大规模数据集的Orb-v3和MACE-OMOL模型也表现出显著优势。
然而,研究同时发现架构设计同样重要。即使拥有更多参数和更大训练集,不同架构之间仍然存在明显性能差异。例如MACE架构在参数利用效率方面普遍优于FeNNix架构。
因此,模型性能并不仅仅由规模决定,网络结构设计仍然是影响最终效果的重要因素。
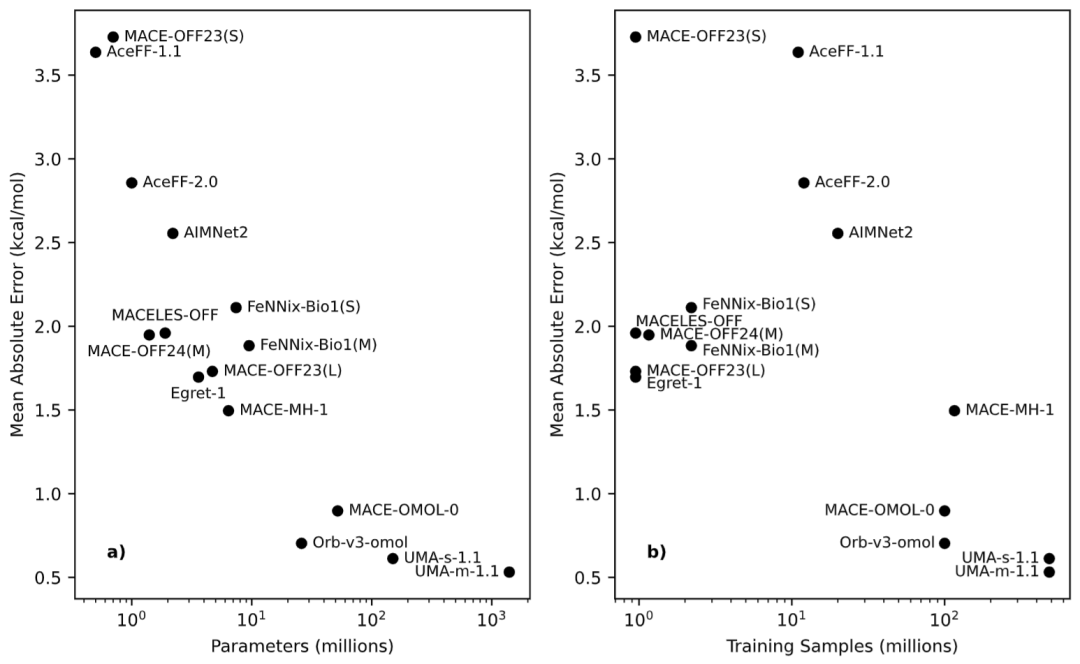
图2: 模型精度与模型规模关系分析。
显式库仑项未表现出明显优势
近年来,许多机器学习势能尝试通过引入显式库仑能项来增强长程相互作用建模能力。研究比较了包含和不包含1/r库仑项的模型后发现,这一策略并未带来明确优势。
对于带电体系预测而言,虽然部分含库仑项模型表现良好,但另一些同类模型误差依然较高。而在大分子体系上的尺度泛化能力方面,同样未观察到一致改善。更值得注意的是,本研究中整体性能最好的多个模型均未采用显式库仑项设计。
研究人员因此认为,目前尚无充分证据证明显式长程库仑项能够系统提升基础势能模型性能,其实际价值仍需进一步研究验证。
不同模型在速度和显存消耗方面差异巨大
除了预测精度之外,研究还系统分析了计算效率。结果显示,不同模型之间的速度差异可达到数十倍以上。在小分子体系中,AIMNet2和FeNNix-Bio1表现最快,而在大规模水盒体系中,FeNNix系列和AceFF系列保持较高计算效率。
对于超大体系模拟,部分高精度模型速度明显下降。例如UMA-m-1.1虽然具有最高精度,但在大型体系中的模拟速度远低于Orb-v3和FeNNix模型。显存需求同样呈现巨大差异。研究发现,显存占用与模型参数规模并非简单线性关系,而更受模型架构影响。有些参数规模巨大的模型仍能处理超过两万个原子的体系,而某些较小模型则在几千个原子规模时就已耗尽显存。
这些结果表明,模型规模并不是决定实际应用能力的唯一因素,高效架构设计同样至关重要。
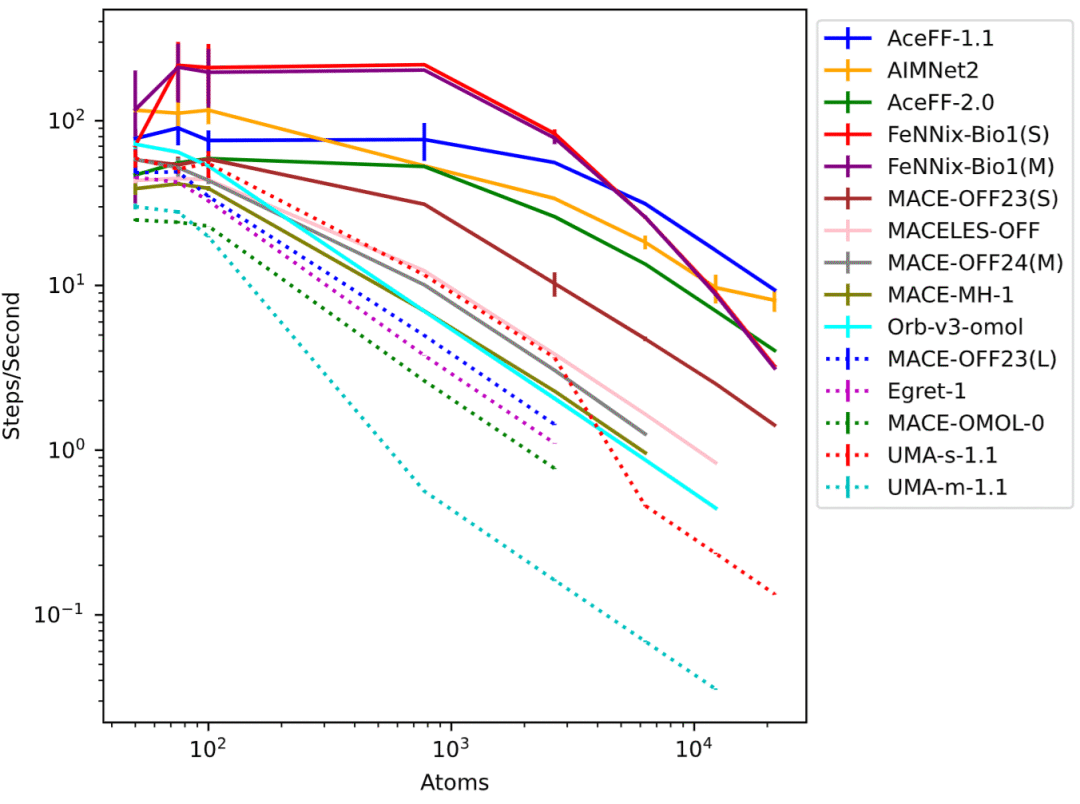
图3: 不同体系规模下的模拟速度比较。
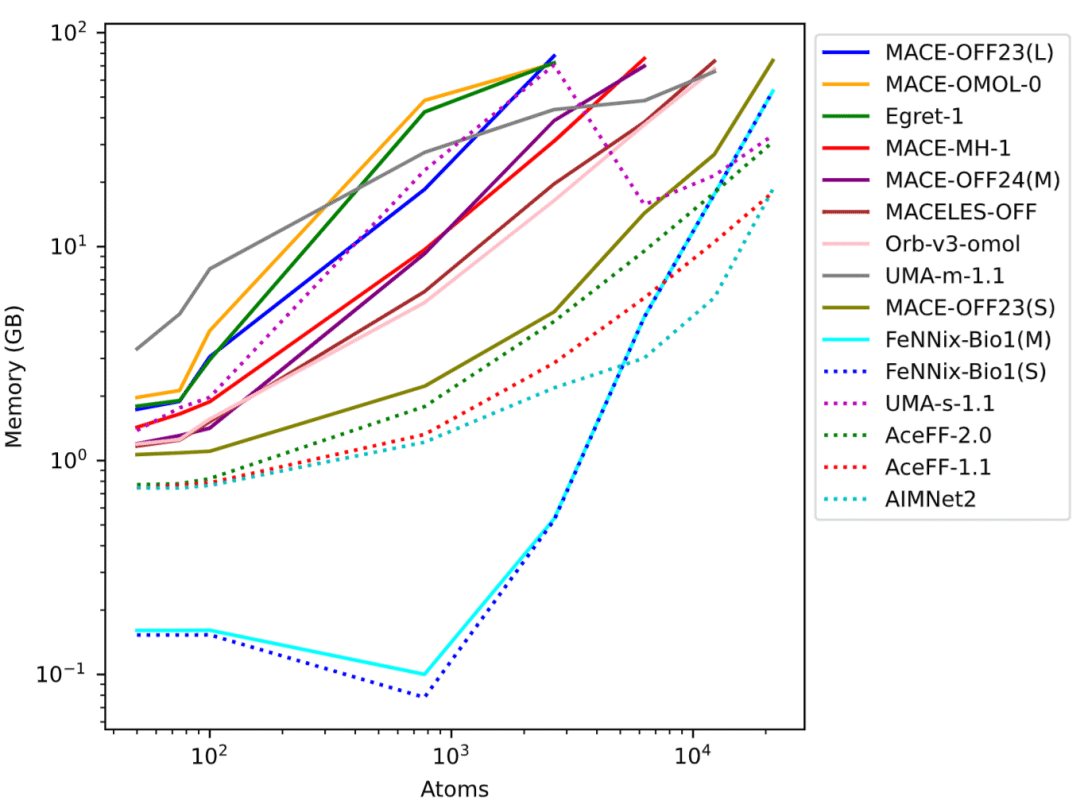
图4: 不同体系规模下的显存占用情况。
精度与效率之间存在明显权衡关系
研究最后分析了精度与效率之间的关系。总体而言,更高精度通常意味着更高计算成本,但这种关系并非绝对。
例如,Orb-v3和UMA-s-1.1在保持高精度的同时仍具有较好的计算效率,因此位于性能前沿。而部分模型虽然计算较慢,却并未获得相应精度提升。
研究利用速度—误差二维空间构建性能前沿分析,识别出在特定精度水平下最具竞争力的模型组合。
对于需要最高精度的应用,UMA-m-1.1是最佳选择;对于兼顾精度和效率的场景,Orb-v3和UMA-s-1.1更具优势;而对于大规模高通量模拟,FeNNix-Bio1和AIMNet2则具有明显速度优势。
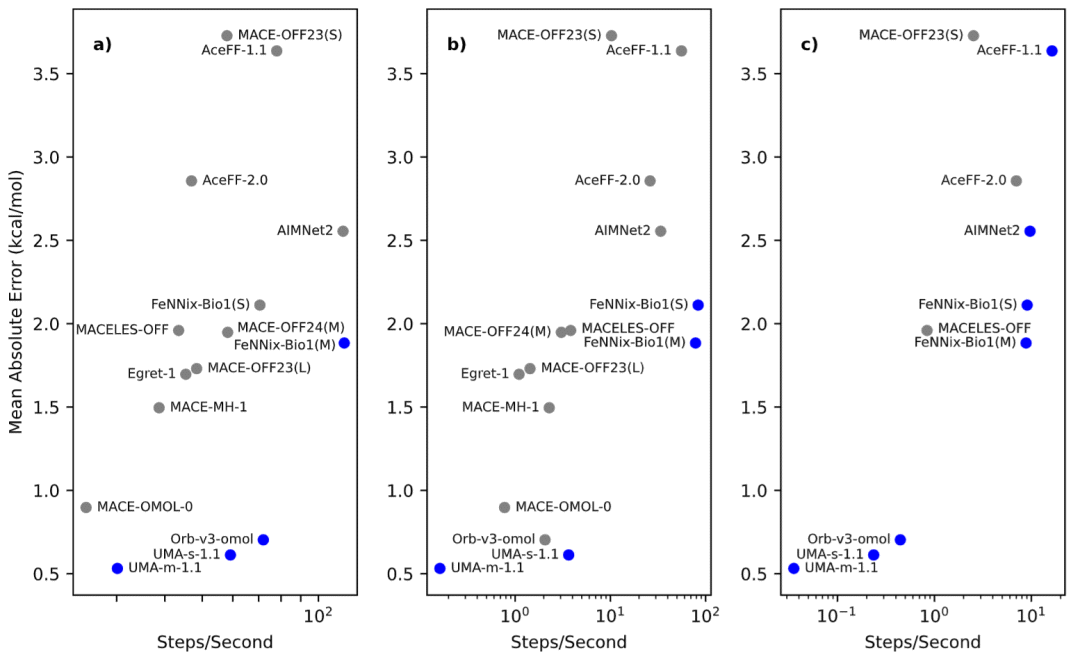
图5: 精度与计算效率权衡分析。
讨论
本研究首次在统一测试框架下系统比较了15种主流预训练机器学习势能模型,为当前快速发展的MLIP领域提供了客观参考标准。
研究结果显示,基础势能模型已经能够在多个分子体系上达到接近化学精度的水平。其中UMA-m-1.1、UMA-s-1.1和Orb-v3-omol是目前最接近“通用分子势能”的代表模型,在准确性方面明显领先于其他方法。
与此同时,研究也揭示了当前MLIP发展的重要规律。模型参数规模和训练数据规模仍然是决定预测性能的核心因素,这与大语言模型和多模态基础模型的发展趋势高度一致。未来更大规模的数据集和模型可能进一步提升精度上限。
然而,研究同样表明,未来发展方向不能仅仅依赖模型扩张。随着精度逐渐接近量子化学参考水平,计算效率将成为更加关键的竞争指标。研究人员认为,未来机器学习势能的发展应更加关注“单位计算成本下的精度提升”,而不仅是单纯追求最低误差。
此外,研究未发现显式库仑长程项能够稳定提升模型性能,这一结果对当前机器学习势能设计理念提出了新的思考。未来仍需进一步研究长程静电相互作用在基础势能模型中的最佳表示方式。
总体而言,本研究表明预训练机器学习势能已经进入基础模型时代。随着OMol25、OMat24等超大规模数据集持续扩展,以及UMA、Orb、MACE等新一代基础势能不断发展,机器学习势能有望逐步成为分子模拟、药物设计和材料发现领域的新一代通用计算引擎。
整理 | DrugOne团队
参考资料
Eastman, P., Pretti, E., & Markland, T. E. (2026). Accuracy and Efficiency Benchmarks of Pretrained Machine Learning Potentials for Molecular Simulations. Journal of Chemical Theory and Computation.

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-17,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号