Acc. Chem. Res. | 抗生素的黄昏,抗菌肽的黎明:AI如何在天然界挖掘、从零开始生成

Acc. Chem. Res. | 抗生素的黄昏,抗菌肽的黎明:AI如何在天然界挖掘、从零开始生成

MindDance
发布于 2026-06-05 20:09:20
发布于 2026-06-05 20:09:20
原文:AI-Driven Antimicrobial Peptide Discovery: Mining and Generation 期刊:Accounts of Chemical Research,2025,58,1831−1846 DOI:10.1021/acs.accounts.0c00594
一、一场正在逼近的全球危机
先说一个让人不安的数字。
到 2050 年,抗微生物耐药性(antimicrobial resistance,简称 AMR)有可能取代癌症,成为人类的头号死因之一。这不是危言耸听的科幻设定,而是写在多份全球卫生报告里的严肃推演。
更糟的是供给端的停滞。20 世纪是抗生素的黄金时代,新药一个接一个被发现。但过去三十年,人类几乎陷入了一段创新的真空期——没有任何全新结构的抗生素类别真正走向市场。一边是病原体的耐药机制飞速进化,一边是新药管线接近枯竭,传统的抗生素发现流程往往要耗费数年才能找到一个临床前候选分子。这是一场我们正在慢慢输掉的赛跑。
正是在这样的背景下,抗菌肽 AMP 成为最被寄予厚望的下一代抗菌武器之一。而这篇发表于《Accounts of Chemical Research》的综述,讲的就是人工智能如何从两个完全不同的方向切入,重新定义抗菌肽的发现——一条路是在浩瀚的天然序列里挖掘,另一条路是从数据分布中凭空生成。

二、为什么是抗菌肽
要理解 AI 在这里能做什么,得先理解抗菌肽到底是一类什么样的分子。
抗菌肽通常很短,长度在 10 到 100 个氨基酸之间,带有净正电荷(常见范围是 +2 到 +9),并且含有较高比例(一般不低于 30%)的疏水氨基酸。这套物理化学特征并非偶然,而是和它的杀菌方式直接绑定的。
带正电的抗菌肽,对带负电的微生物细胞膜有天然的选择性亲和力,却不太会去攻击真核细胞那层电中性的膜。换句话说,它在设计上就倾向于敌我分明。除了直接破坏膜结构,抗菌肽还能通过抑制蛋白质或核酸合成、干扰蛋白酶活性、阻断细胞分裂等多种机制来杀灭微生物。按照氨基酸组成和结构,抗菌肽还可以细分为富含脯氨酸、富含色氨酸与精氨酸、富含组氨酸或甘氨酸等不同类别,多数呈 α 螺旋构象,也有 β 折叠、线性延伸或混合构象。从哺乳动物、两栖类、昆虫到微生物,整个生命之树上都能找到它们的身影,它们是宿主对抗病原体感染的天然防线。
抗菌肽最吸引人的一点是:微生物对它产生耐药性的速度,远比对传统抗生素来得慢。这正是它有望跳出耐药困局的关键。
但事情没那么简单。许多抗菌肽对哺乳动物细胞同样有毒性,通常用溶血活性和细胞毒性来衡量。再加上已知抗菌肽在临床上的成功案例相当有限,于是核心难题被清晰地勾勒出来——
我们需要的,是既高效杀菌、又对人体足够安全的肽。设计抗菌肽,本质上就是在活性与毒性之间寻找平衡。
三、一场近乎不可能的搜索
如果把抗菌肽设计看成一道数学题,它会是这样一道令人绝望的优化问题:在一个极其庞大的潜在序列空间里,找出最有活性、毒性又最低的那一小撮肽,而我们手头关于这些肽性质的数据却少得可怜。
到底有多庞大?文章给了一个直观的算账。仅考虑长度不超过 25 个氨基酸的肽,如果用暴力穷举去逐一评估,需要处理的序列数量大约在 10 的 32 次方 这个量级——这在计算上是彻底不可行的。
而作为对照,经过实验验证的抗菌肽(基于 DBAASP 数据库估算)只有大约 10 的 4 次方个;如果再限定到针对某个特定细菌(比如大肠杆菌)有明确活性记录的,更是只剩下 10 的 3 次方左右。已知与未知之间,隔着天文数字般的鸿沟。
要在如此广阔的空间里高效导航,就必须依靠足够聪明的算法。而所有算法都要在两种取向之间做权衡,作者把它概括为一组很漂亮的对立——现实主义与理想主义的权衡(realism-idealism trade-off):
- 现实主义,意味着选择那些与现有抗菌肽相似、因而更可能真实存在、可被天然合成的序列;
- 理想主义,意味着大胆优化,追求超高活性和超低毒性,哪怕代价是可能造出现实中并不存在的怪东西。
这组对立,恰好对应了 AI 驱动抗菌肽发现的两条主线:偏向现实主义的挖掘,与偏向理想主义的生成。
四、全局地图:判别、挖掘、生成
在深入两条主线之前,先看一张总览图。下面这张图来自原文,把整个 AI 驱动抗菌肽发现的框架浓缩在了一起。
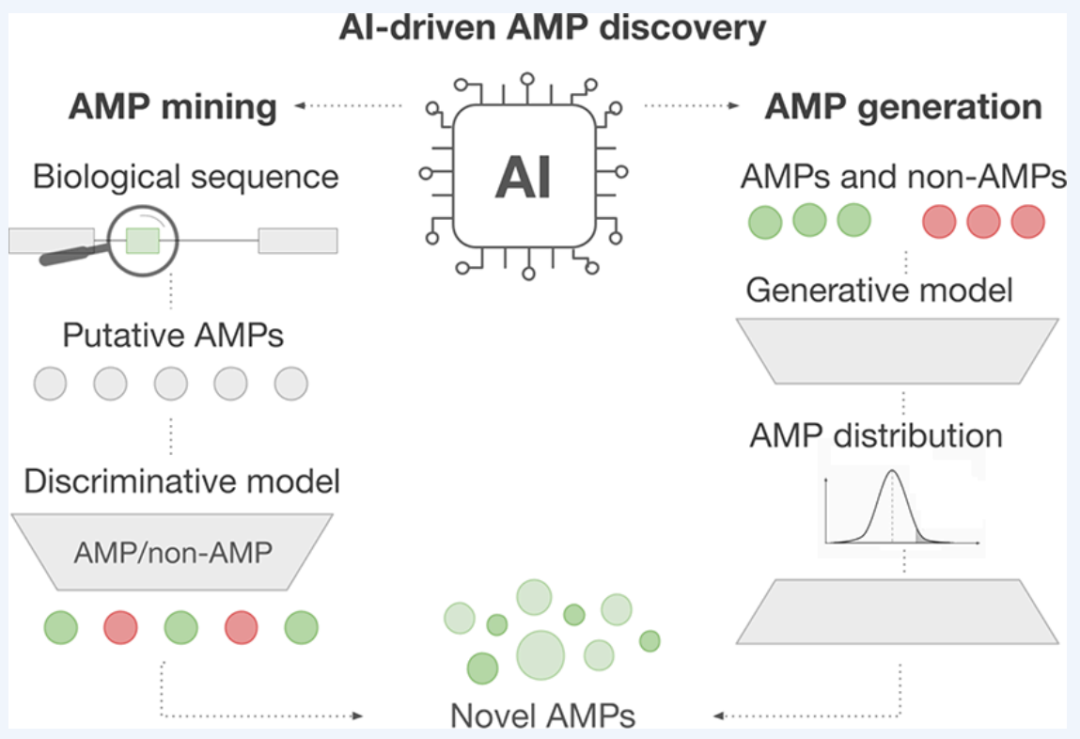
AI 驱动的抗菌肽发现总览:左侧为挖掘路径,右侧为生成路径,两者最终汇聚到新型抗菌肽
AI 驱动的抗菌肽发现总览:左侧为挖掘路径,右侧为生成路径,两者最终汇聚到新型抗菌肽
读懂这张图,也就读懂了全文的骨架。整套体系由三件工具构成:
- 判别式方法——贯穿始终的工具箱,负责预测一条肽到底有没有活性、有没有毒性,是挖掘和生成共同依赖的裁判;
- 挖掘(左侧)——把判别模型用在天然的基因组、蛋白质组、宏基因组序列上,把藏在生命之树里的抗菌肽筛出来;
- 生成(右侧)——让生成模型学会抗菌肽数据的分布,然后创造出训练集里从未出现过的全新序列。
下面,我们一件一件来看。
五、工具箱:判别式模型,AI 的裁判席
无论是挖掘还是生成,最后都绕不开同一个问题:眼前这一大堆候选肽,哪些真的有用?回答这个问题的,就是判别式模型。它的任务大致有三类——区分抗菌肽与非抗菌肽、预测活性(比如最低抑菌浓度 MIC)、预测毒性。
按照所用的算法范式,判别模型大致经历了三代演进。
第一代是传统机器学习。 决策树、支持向量机(SVM)、随机森林(RF)这些方法完全依赖从序列里手工提取的描述符。别看它们简单,在某些任务上的表现并不输给复杂的深度学习模型。其中一个很有代表性的例子是 Macrel,一个随机森林模型。它特意用了大约 1∶50 的抗菌肽与非抗菌肽比例来训练——这个不平衡的设计,是为了更真实地模拟基因组挖掘时的实际分布,而不是用一个理想化的平衡数据集自欺欺人。后来在大规模挖掘项目 AMPSphere 中,Macrel 就立了大功。
第二代是深度学习。 有意思的是,抗菌肽预测里最主流的深度学习架构,大多是从自然语言处理借来的——循环神经网络(RNN)、长短期记忆网络(LSTM),以及后来无处不在的注意力机制。这背后的逻辑是:生物序列和自然语言一样有它的语义,模型通过分析氨基酸的出现频率、前后依赖关系,就能读懂这套语义。比如 AMPlify 结合了双向 LSTM 和注意力层;MBC-Attention 用多分支 CNN 加注意力来回归抗菌肽对大肠杆菌的 MIC;AMPpred-MFA 则同时调动双向 LSTM、CNN 和多头注意力。甚至还有人提出用量子支持向量机来预测肽的毒性。
第三代是大语言模型与蛋白质语言模型(PLM)。 当 ChatGPT 们大放异彩,基于 Transformer 的大模型也被引入了这个领域。把语言模型用在蛋白质序列上,就得到了所谓的蛋白质语言模型。它的训练通常分两步:先在海量蛋白质语料上做生成式预训练,再针对具体的下游任务(功能、性质、结构预测)做微调。这里有一个值得划重点的细节——
肽比典型蛋白质更短、结构更简单,已知的生物活性肽数量也远少于蛋白质。如果直接把在蛋白质上预训练的模型拿来用、不做额外微调,模型就会带着浓重的蛋白质偏见,无法准确刻画肽的分布。
所以,针对肽和抗菌肽特点的额外预训练或微调,往往能带来实打实的提升。此外,预训练语料的选择也很关键——用多样性更高的语料(比如序列间相似度更低的 UniRef50,相比 UniRef100),哪怕架构完全不变,效果也会更好。常见的语言模型架构包括 BERT 系列以及融合了序列与进化信息的 ESM 编码器。
至于肽的表示方式,模型也各有讲究。最常见的当然是氨基酸序列本身,但也可以转化为序列描述符、预训练模型的嵌入向量,甚至把序列转成图像后用 CNN 处理。还有一类很有意思的尝试是引入结构信息:sAMP-pred-GAT 用图注意力网络整合结构、序列和进化信息;AMPredictor 则是个图卷积网络,结合了分子指纹、肽的接触图和 ESM 嵌入来回归 MIC。
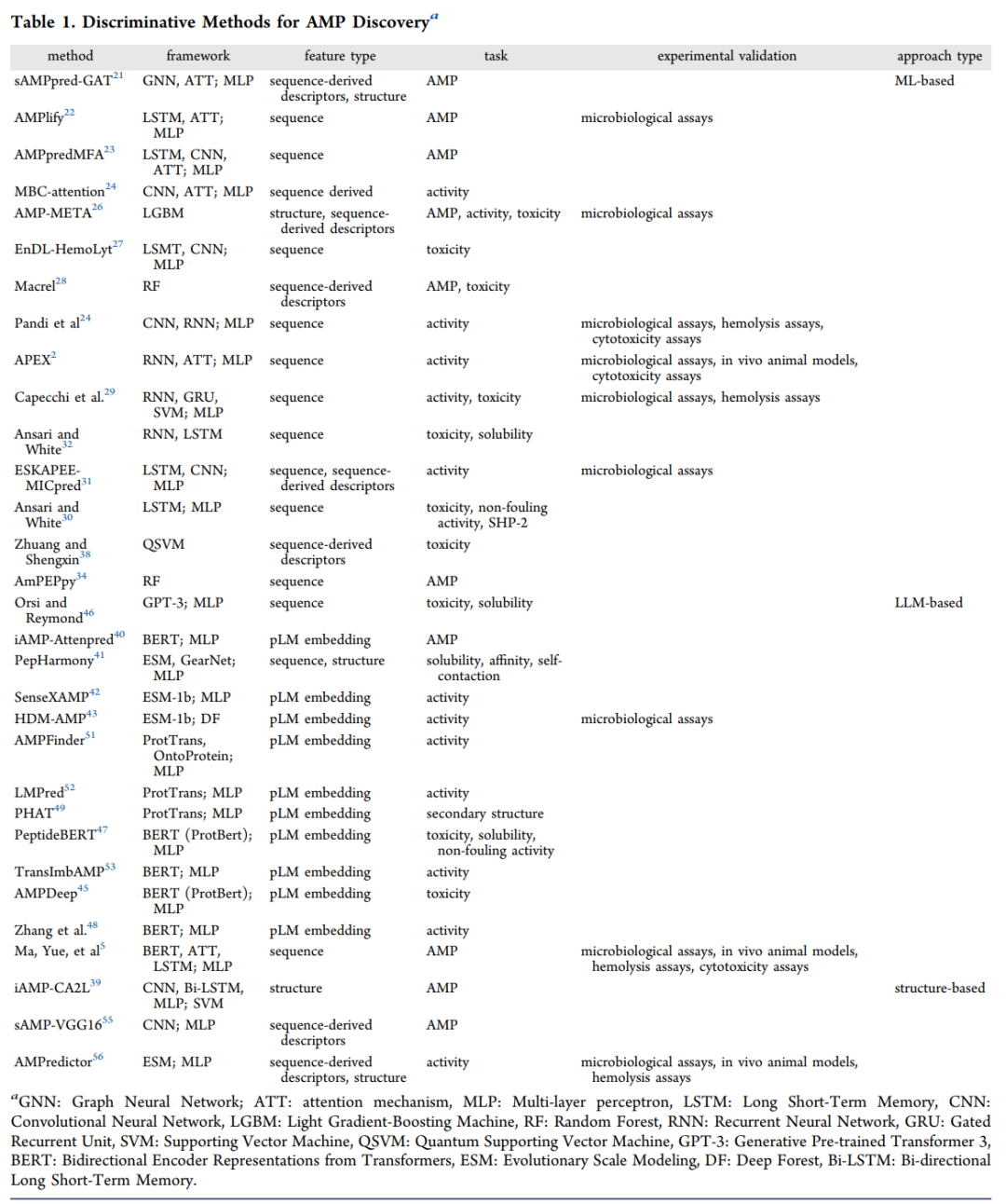
原文用上面这个表系统梳理了近年的判别方法,涵盖框架、特征类型、任务与实验验证情况。下面是按范式整理的一个浓缩版,方便快速建立印象:
范式 | 代表方法 | 核心架构 | 主要任务 |
|---|---|---|---|
传统机器学习 | Macrel、AmPEPpy | 随机森林 | 抗菌肽判别、毒性 |
深度学习 | AMPlify、MBC-attention、APEX | LSTM/CNN/注意力 | 判别、活性 |
蛋白质语言模型 | PeptideBERT、SenseXAMP、HMD-AMP | BERT/ESM | 活性、毒性、溶解度 |
结构图模型 | sAMP-pred-GAT、AMPredictor | 图神经网络 | 判别、活性 |
不过,作者也点出了一个令人警醒的现实:在所有这些判别模型里,真正经过微生物学实验验证的只是少数,而经过溶血和细胞毒性实验验证的就更少了。算法很热闹,湿实验却跟不上——这个矛盾,后文还会反复出现。
六、路径一:挖掘——在生命之树里寻宝
挖掘的思路朴素而强大:既然天然界里本来就有抗菌肽,那就把判别模型当成探测器,到基因组、蛋白质组、宏基因组这些海量序列里去把它们筛出来。
这条路的最大优点在于真实。天然来源的肽只含 L 型氨基酸,可以用固相合成法低成本、低副产物地合成出来;而那些在肠道微生物组等环境中分泌的抗菌肽,本来就要在活体里精准打击入侵微生物、同时避免伤害宿主——这意味着它们天生就在活性与安全之间做过权衡。值得一提的是,多数挖掘方法并不依赖毒性预测器,原因恰恰是当前毒性预测的可靠性还不够。
挖掘能成功,有两个前提:第一,被挖掘的序列里确实埋着高活性、低毒性的抗菌肽;第二,所用的判别模型确实有本事把它们认出来。这两点决定了挖掘既要有富矿,也要有好探头。
富矿:可供挖掘的序列宝库
今天,数以百万计的基因组已经可以公开获取,连同宏基因组(包含一个微生物群落中多个生物的遗传物质)和蛋白质组一起,被收录进各种公共数据库。比如全球微生物基因目录 GMGCv1,由成千上万个跨越不同生境的宏基因组构建而来,包含数十亿个开放阅读框,聚类后得到数以亿计的物种级基因。另一个例子是 Duan 等人构建的全球微生物小开放阅读框目录 GMSC,收录了接近一百万条非冗余的小开放阅读框,并配套了名为 GMSC-mapper 的工具用于识别和注释小蛋白。
挖基因组与蛋白质组
人类自己,就是一座金矿。 Torres 等人的一项标志性研究,用一套基于关键物理化学性质(序列长度、净电荷、平均疏水性)的算法去预测抗菌活性,扫描了人类蛋白质组中的 42,361 条蛋白序列,识别出 2,603 个潜在的抗菌肽候选。这些候选中有许多此前从未被认作抗菌分子,甚至从未被认为参与宿主免疫。其中若干个被合成出来、做了实验验证,并在动物模型中展现了疗效。这项工作的妙处在于,它刻意绕开了已知的抗菌肽序列模体,只盯着物理化学特征,反而挖到了真正新颖的抗菌分子。
最浪漫的,是向灭绝的物种借药。 AI 还把挖掘的触角伸向了已经消失的生命——尼安德特人、丹尼索瓦人,乃至披毛犸象。这门崭新的领域被称为分子去灭绝(molecular de-extinction)。在相关工作中,研究者一边引入了用于全蛋白质组切割位点预测的随机森林模型 panCleave,一边动用了更强大的深度学习模型 APEX,从所有可获取的灭绝生物蛋白质组里,发掘出诸如尼安德特素-1、猛犸象素-2、象素-2 等全新抗菌肽,它们如今已是临床前候选分子。
这些计算努力,把一个曾经需要数年的发现过程,压缩到了数小时之内。
噬菌体也没被放过。 Wu 等人针对 ESKAPE 病原体(粪肠球菌、金黄色葡萄球菌、肺炎克雷伯菌、鲍曼不动杆菌、铜绿假单胞菌、肠杆菌属这一组临床上极危险的细菌)及其相关噬菌体,搭建了一套挖掘流水线,用 CNN 加 LSTM 训练模型评估活性,最终建成了 ESKtides 数据库,里头收录了 超过 1,200 万条 预测高抗菌活性的肽。
挖微生物组
人类肠道微生物组是另一片沃土。Ma 等人用 LSTM、注意力和 BERT 等深度学习技术,从中挖出了 181 条有抗菌活性的肽,其中很多与已知抗菌肽的序列同源性不足 40%,并在小鼠肺感染模型中有效降低了细菌负荷、对耐药革兰氏阴性菌展现出显著活性。
有意思的是,由于抗癌肽(ACP)和抗菌肽存在重叠,同样的思路被迁移到了抗癌肽预测上:一项研究从肠道宏基因组里识别出 40 个潜在抗癌肽,其中 39 个在多种癌细胞系中显示出抗癌活性,更有 2 个能在小鼠模型中显著缩小肿瘤且不致毒。
而最大规模的一次挖掘,发生在整个全球微生物组。 Santos-Júnior 等人用机器学习分析了 63,410 个宏基因组和 87,920 个微生物基因组,并用蛋白质组学与转录组学数据作为基因组识别之后的过滤步骤,最终计算预测出 近百万个 新抗菌肽候选,悉数存入了 AMPSphere 数据库。另一项整合了人体四个部位宏基因组的研究,识别出 323 个候选抗菌肽,在体外和体内都对临床相关病原体显示出活性。除人体之外,还有研究把目光投向能维持微生物组稳态的特殊物种,比如淡水水螅 Hydra 的微生物组和蟑螂肠道微生物组。

原文的上面这个表完整列出了这些挖掘工作的工具、模型、序列类型与来源。下面是一个精选版,串起几条最有代表性的故事线:
研究 / 工具 | 序列来源 | 模型 | 关键成果 |
|---|---|---|---|
Torres 等人 | 人类蛋白质组 | 物理化学性质打分 | 42,361 条序列中识别 2,603 个候选 |
APEX(分子去灭绝) | 灭绝物种蛋白质组 | RNN+注意力 | 尼安德特素-1、猛犸象素-2、象素-2 |
Ma 等人 | 人类肠道微生物组 | LSTM+注意力+BERT | 181 个活性肽,小鼠肺感染有效 |
Santos-Júnior 等人 | 全球微生物组 | 随机森林(Macrel) | 近百万候选,建成 AMPSphere |
ESKtides(Wu 等人) | ESKAPE 噬菌体 | CNN+LSTM | 1,200 万+预测高活性肽 |
不挖天然序列,那就穷举短肽
还有一条剑走偏锋的路:与其去天然序列里淘金,不如干脆把某个固定短长度下的所有可能序列全部评估一遍。Huang 等人就开发了一套机器学习流水线,系统扫描了由 6 到 9 个氨基酸构成的庞大虚拟肽库,通过多个串联模块去过滤、分类、排序、预测候选肽的效力。由于他们的判别模型训练自 GRAMPA 数据集(一个汇编的 MIC 测量集合,可能存在实验室特异性偏差),他们特意采用了两步实验验证策略,在第一阶段后再去精修判别器以减小偏差。最终成果包括 3 个强效六肽,对多重耐药病原体有强抗菌活性,在小鼠感染治疗中疗效可与青霉素媲美,且毒性很低。
另一项专门针对鲍曼不动杆菌的研究,扫描了六肽、七肽、八肽的全部文库,候选数量高达数百亿。更惊人的是,它的分类器只用了 148 条 极度稀缺的训练序列,靠的是一套包含预训练和多次微调的小样本学习策略。
七、路径二:生成——从零创造一条新肽
如果说挖掘是淘金,那么生成就是炼金。生成式 AI 的逻辑是:通过学习和建模数据背后的分布,直接产出训练集中从未出现的全新肽序列,并在生成的同时向着高活性、低毒性的方向去优化。这条路追求的是理想主义——它有机会造出超越天然的合成肽,但也冒着造出不够真实序列的风险。
用什么框架生成
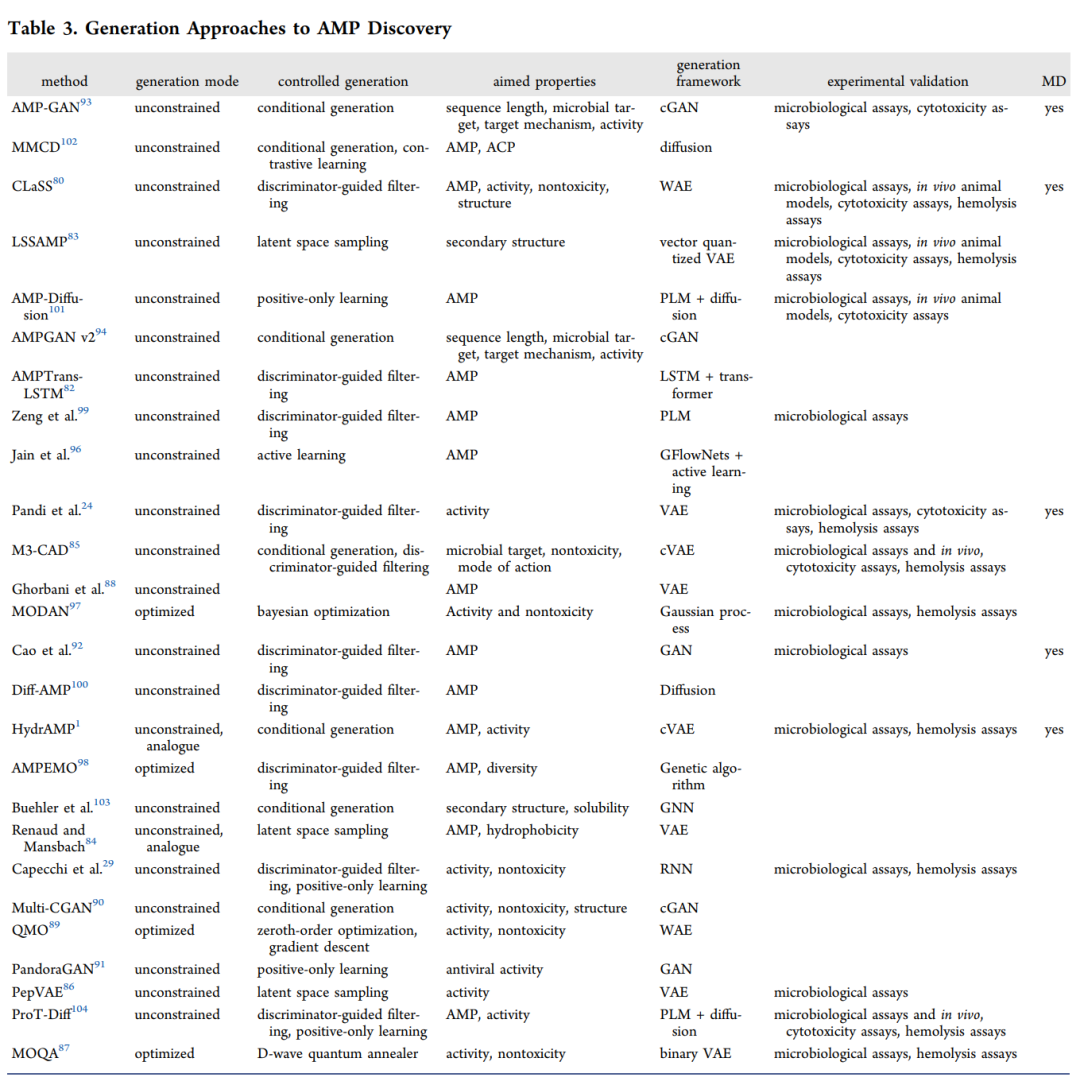
同样,上面这个表系统梳理了各类生成方法及其框架。总体来看,自回归模型(LSTM、RNN)虽然被尝试过,但目前用得相对较少;研究主力集中在变分自编码器(VAE),其次是 Wasserstein 自编码器(WAE) 和生成对抗网络(GAN),扩散模型也在快速兴起。一个不得不承认的现实是:虽然不少生成研究做了微生物学测试,但真正进一步在动物模型中验证的还很少。
关键难题:如何把生成引向我们想要的方向
生成模型能在短时间里吐出成千上万条貌似合理的候选肽,所以真正的挑战是——如何引导这个过程,让产物更可能具备我们想要的性质。文章梳理了几种主流的控制策略:
- 判别器引导过滤。 用一个辅助判别器去引导生成、筛选顶尖候选。比如 CLaSS 模型,就在 WAE 的潜空间上训练判别器,把生成引向目标活性和毒性。
- 仅正样本学习。 像 PandoraGAN 那样,只用高活性的肽来训练。
- 条件生成。 GAN 与 VAE 演化出了条件变体——条件 GAN(cGAN)和条件 VAE(cVAE),在生成阶段就配置好目标条件。Multi-CGAN 可以同时优化多个性质;M3-CAD 则是一个多模态、多任务、多标签的 cVAE,一口气瞄准包括预测三维结构、物种特异性活性、抗菌机制、毒性在内的八类特征。
- 潜空间采样。 直接从潜在空间里那些被认为编码了理想属性的区域去取样。比如 LSSAMP,把潜在表示离散化,同时编码序列和结构信息,从而生成具有特定二级结构的肽。
但作者也诚实地指出:判别器引导过滤和仅正样本学习,都受制于同一个瓶颈——正样本(既有活性又无毒的抗菌肽)实在太稀缺了。
主角登场:HydrAMP
作为这篇综述作者团队自己的代表作,HydrAMP 被着重介绍,它针对标准 cVAE 框架做了几项关键改进,专门用来对付训练数据匮乏的难题。
它通过对低 MIC 值等性质进行条件约束,专注于生成高活性抗菌肽;内置一个预训练分类器,确保生成的肽保留住目标性质;为提升训练稳定性,作者在损失函数里加了几项,既保证生成的肽与输入足够接近,也保证输入与生成肽在潜空间里的表示彼此匹配。HydrAMP 最有特色的能力,是可以修改一条已有的肽去满足特定活性条件,而这个过程由一个创造力参数(creativity parameter)来调控——创造力越高,产生的类似物越多样。和只会从潜空间采样的标准 cVAE 不同,HydrAMP 既能改良已知抗菌肽,也能把那些实验证明本无抗菌活性的肽点石成金。
在筛选环节,分子动力学(MD)模拟提供了额外的活性描述符,与分类器集成一起为候选肽排序。最终,最有希望的肽被合成出来、做活性和毒性的实验验证。借助 HydrAMP,作者团队发现了 15 个全新的强效抗菌肽,对包括多重耐药菌株在内的多种细菌都有活性。
更进一步:直接优化与语言模型
除了像 HydrAMP 那样的类似物生成,另一个面向理想主义肽设计的方向是直接优化生成,常配合定制的代价函数。这一领域里,QMO 用零阶梯度优化来导航潜空间;其他应对优化挑战的方法还包括基于 GFlowNets 的主动学习、量子退火、贝叶斯优化以及进化算法。
至于大语言模型,虽然它们在判别任务中已被广泛使用,但在生成任务里的应用还相对有限。从蛋白质语言模型做生成,通常要么用类似 GPT 的解码器架构,要么在预训练模型给出的连续嵌入上跑一个扩散过程。这些方法目前的控制策略还比较简单,多依赖仅正样本学习或判别器过滤。一个有前景的方向是对比学习,比如 MMCD,在训练扩散模型时把已知正样本抗菌肽的嵌入与负样本进行对比。
下面是生成路径的一个精选概览:
方法 | 生成框架 | 控制策略 | 关键特点 |
|---|---|---|---|
HydrAMP | 条件 VAE | 条件生成 | 创造力参数、类似物生成、15 个强效新肽 |
CLaSS | Wasserstein 自编码器 | 判别器引导过滤 | 在潜空间训练判别器 |
M3-CAD | 条件 VAE | 条件生成 | 多模态多任务,瞄准八类性质 |
Multi-CGAN | 条件 GAN | 条件生成 | 多性质同时优化 |
QMO | Wasserstein 自编码器 | 零阶优化 | 潜空间梯度优化 |
MMCD | 扩散模型 | 对比学习 | 正负样本嵌入对比 |
八、两条路径的对照
读到这里,挖掘与生成的分野已经很清晰了。它们不是竞争关系,而是从两个不同哲学出发、各有所长的互补策略。一张表说清楚:
维度 | 挖掘 | 生成 |
|---|---|---|
核心思路 | 在天然序列里筛选已存在的肽 | 学习分布后创造全新序列 |
哲学定位 | 现实主义 | 理想主义 |
输出特点 | 天然存在、只含 L 型氨基酸、易合成 | 全新、有望超越天然 |
主要方法 | 判别模型扫描基因组/蛋白质组 | VAE/GAN/扩散模型 |
主要风险 | 受限于天然序列本身的质量 | 可能生成不真实的序列 |
体内验证 | 相对较多 | 相对较少 |
值得注意的是,无论走哪条路,判别式模型都站在裁判席上——挖掘靠它筛选,生成靠它引导和过滤。三件工具其实是一个有机的整体。
九、仍未跨越的鸿沟
这篇综述最可贵的地方,是它没有停留在歌颂技术,而是用相当大的篇幅冷静盘点了三个层面尚未解决的难题。
判别式模型的难题。 首先是老生常谈但极其要命的数据稀缺——尽管迁移学习和预训练大模型能部分缓解,但针对多重耐药菌株的数据,恐怕在很长时间里都不足以训练出菌株特异的活性预测器。其次是缺乏经过验证的负样本:没人有动力去发表阴性数据,而一条肽也可能因为没对敏感菌株做测试、或因为溶液中结块等技术问题被误判为无活性;研究还表明,负样本数据集的构建方式会严重影响模型表现。再者,毒性预测明显落后——溶血活性预测表现欠佳,细胞毒性预测更是严重缺位,而毒性恰恰是抗菌肽走向临床的主要障碍。此外,结构信息利用不足、模型很少在独立外部数据集上被客观评估、无法处理含有环化、β 氨基酸、修饰半胱氨酸、脂质连接等非经典结构的修饰肽(而像多黏菌素这样已获 FDA 批准的抗菌肽恰恰是修饰肽)——这些都限制了判别方法在临床相关抗菌肽上的适用性。半衰期、ADMET 等关键性质的训练数据,目前也几乎是空白。
挖掘的难题。 挖掘的成败,取决于既要有富矿、又要有好探头这两个前提;判别模型的局限会自然传导到挖掘上——既然判别模型认不出复杂的化学修饰肽,那么当前能挖出来的就只有线性肽。基因组上下文这一对功能预测至关重要的信息尚未被充分利用,把转录组、核糖体测序等更多数据类型整合进来、利用生物合成基因簇、分析多序列比对而非单条序列,都是可能的增益方向。一个必须正视的事实是:尽管近期的挖掘研究确实在动物模型里做了临床前测试,但还没有任何一个候选分子真正进入临床研究。
生成的难题。 生成模型的评估与基准测试本身就很难——生成的肽通常只能用多样性、新颖性、与训练数据的相似度来评价,而真正的活性和毒性除了极小的验证子集外都是未知;不同研究选用的辅助判别器又完全是任意的,导致模型之间根本无法公平比较。如何从成千上万的候选里高效排序也是难题,目前主要靠大量过滤加专家经验。更根本的限制在于,绝大多数生成方法只在 20 种氨基酸的字母表上运作,无法采样翻译后修饰和非标准氨基酸所撑开的庞大空间,因而严重低估了真实肽世界的复杂度。和挖掘相比,生成路径在体内得到验证的肽更少,这或许是因为多数 AI 实验室的湿实验能力有限——而这恰恰呼唤 AI、化学、生物实验室与产业伙伴携手,去打通从发现到临床的完整链条。
十、END:从数年到数小时
把视角拉远,这篇综述其实在讲一件正在发生的事:抗菌肽的发现,正站在一场变革的门槛上。
自从早期的开创性工作证明机器能够设计出在小鼠模型中有效的肽类抗生素以来,这个领域已经显著成长与成熟。AI 驱动的方法,已经实实在在地把我们识别新抗菌肽的能力大幅加速——一个曾经以年计的过程,如今可以在数小时内完成。借助大规模的基因组与蛋白质组数据,再配合愈发精巧的生成式与判别式模型,AI 有望针对那些不断涌现的耐药病原体,量身设计出强效的抗菌肽。
当然,从计算预测到临床应用,中间还隔着数据、毒性、修饰肽、体内验证等一道道现实的鸿沟。但方向已经清晰:挖掘负责守住现实主义的底线,去发现天然界已经准备好的答案;生成负责打开理想主义的天花板,去创造自然从未给出的可能;而判别模型,则是贯穿两条路径、决定一切的裁判。
在抗生素逐渐失灵的黄昏里,这套人机协同的体系,正在为抗菌肽点亮一个值得期待的黎明。
参考文献
Accounts of Chemical Research, AI-Driven Antimicrobial Peptide Discovery: Mining and Generation
Acc. Chem. Res. 2025, 58, 12, 1831–1846
https://doi.org/10.1021/acs.accounts.0c00594
本文为基于原始论文的深度解读,所有数据与结论均来自上述文献。如需引用具体细节,请以原文为准。

本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-05,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号