scRNA-hdWGCNA共表达网络分析【3】:模块特征差异分析及模块特征相关性

scRNA-hdWGCNA共表达网络分析【3】:模块特征差异分析及模块特征相关性

KS科研分享与服务-TS的美梦
发布于 2026-06-03 20:03:31
发布于 2026-06-03 20:03:31
1简介
前期已经完成了hdWGCNA的核心分析(scRNA-hdWGCNA共表达网络分析【1】:(一文掌握)详细讲解教程更新及网络图可视化函数,scRNA-hdWGCNA共表达网络分析【2】:富集分析及大型球体网络图构建可视化),构建了网络,识别了基因模块,这里对模块进行深入分析,探究模块的生物学意义。加载数据:
library(hdWGCNA)
library(WGCNA)
library(Seurat)
library(tidyverse)
library(igraph)
library(cowplot)
library(patchwork)
library(dplyr)
library(ggplot2)
library(stringr)
setwd('/Users/ks_ts/Documents/公众号文章/hdWGCNA复现/')
seurat_obj <- readRDS('./Agrp_hdWGCNA_obj.rds')2差异模块分析:Differential module eigengene (DME) analysis
前期的分析已经了解了有哪些基因模块,也分析了模块功能,接下来更希望了解差异,例如演示数据的实验设计是性别,营养差异,更希望揭示不同性别和营养状态下条件特异性的模块调控模式。本节演示如何对共表达网络模块进行差异表达分析(Differential Module Eigengene,DME),以揭示在特定细胞群体中上调或下调的模块。hdWGCNA提供FindDMEs 函数用于两组之间差异模块分析,其语法类似于Seurat中的FindMarkers函数。
group1 <- seurat_obj@meta.data %>% subset(sexXnutr == 'F_Fast' & cell_type3 == 'Agrp') %>% rownames
group2 <- seurat_obj@meta.data %>% subset(sexXnutr == 'M_Fast' & cell_type3 == 'Agrp') %>% rownames# 结果是 group1 vs group2
DMEs <- FindDMEs(
seurat_obj,
barcodes1 = group1,
barcodes2 = group2,
harmonized = TRUE,#默认T,我们前面的ME做了harmny矫正
test.use='wilcox',
wgcna_name='ARH'
)head(DMEs)## p_val avg_log2FC pct.1 pct.2 p_val_adj module## Agrp-M10 0.0003634394 0.98248499 0.855 0.676 0.005088152 Agrp-M10## Agrp-M11 0.0004068578 -0.86945801 0.809 0.919 0.005696009 Agrp-M11## Agrp-M6 0.0006566490 -0.35091115 0.800 0.635 0.009193085 Agrp-M6## Agrp-M13 0.0016294910 0.70616560 0.818 0.608 0.022812875 Agrp-M13## Agrp-M4 0.0020561082 0.01432549 0.955 0.865 0.028785515 Agrp-M4## Agrp-M12 0.0031978977 1.30940087 0.891 0.784 0.044770567 Agrp-M12结果可视化:使用PlotDMEsLollipop可视化的时候需要注意一个问题,DMEs module名称要与seurat_obj一致,否则做图有问题。比如这里我们的module名称就发生了改变,seurat_obj中如下:
mod_color_df <- GetModules(seurat_obj)
unique(mod_color_df$module)## [1] grey Agrp_M1 Agrp_M2 Agrp_M3 Agrp_M4 Agrp_M5 Agrp_M6 Agrp_M7
## [9] Agrp_M8 Agrp_M9 Agrp_M10 Agrp_M11 Agrp_M12 Agrp_M13 Agrp_M14
## 15 Levels: grey Agrp_M1 Agrp_M2 Agrp_M3 Agrp_M4 Agrp_M5 Agrp_M6 ... Agrp_M14但是经过FindDMEs之后”_“变成了”-“,以前也解释过这个问题,FindMarkers的毛病。
unique(DMEs$module)## [1] "Agrp-M10" "Agrp-M11" "Agrp-M6" "Agrp-M13" "Agrp-M4" "Agrp-M12"
## [7] "Agrp-M5" "Agrp-M3" "Agrp-M9" "Agrp-M1" "Agrp-M2" "Agrp-M8"
## [13] "Agrp-M7" "Agrp-M14"所以,修改一下即可:
DMEs$module <- gsub("-", "_", DMEs$module)
rownames(DMEs) <- DMEs$module
DMEs可视化:
PlotDMEsLollipop(seurat_obj,
DMEs,
wgcna_name='ARH',
pvalue = "p_val_adj")&
theme_classic()&
labs(title = "F_Fast vs M_Fast")&
theme(plot.title = element_text(hjust = 0.5))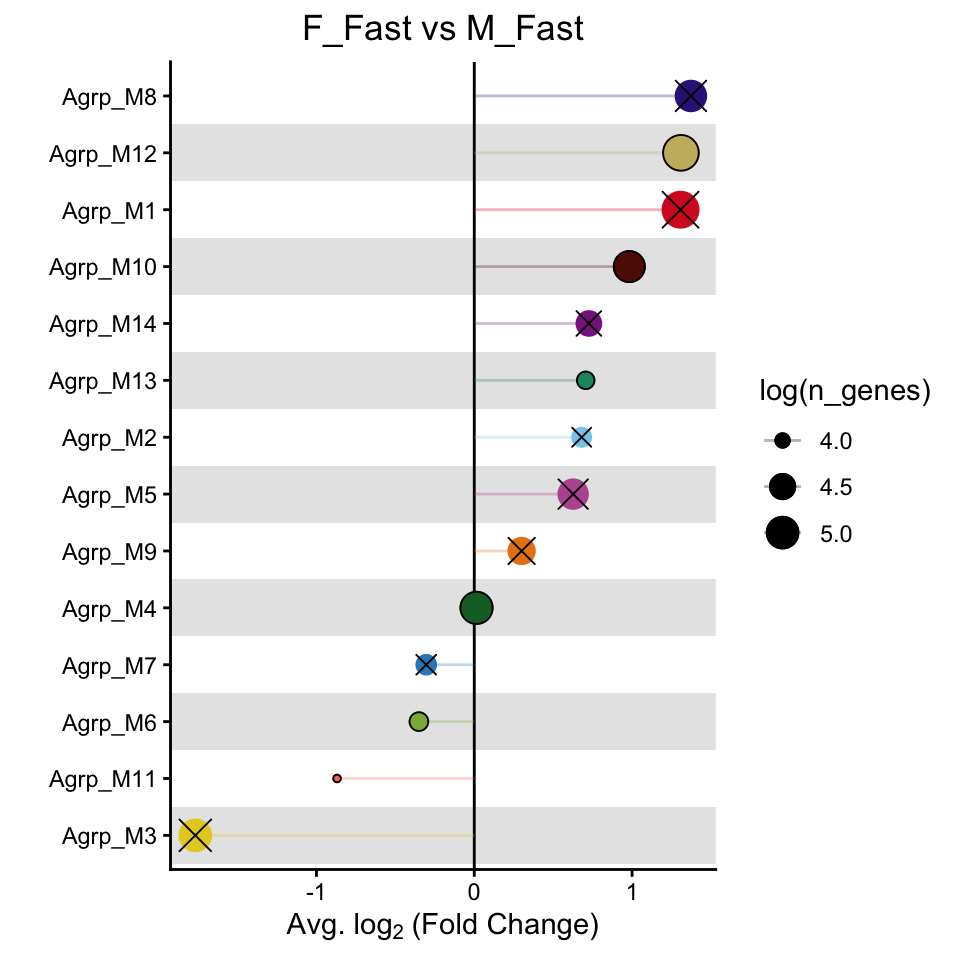
另外一种火山图展示方式可能更明确:
PlotDMEsVolcano(seurat_obj,
DMEs,
wgcna_name = 'ARH')&
labs(title = "F_Fast vs M_Fast")&
theme(plot.title = element_text(hjust = 0.5))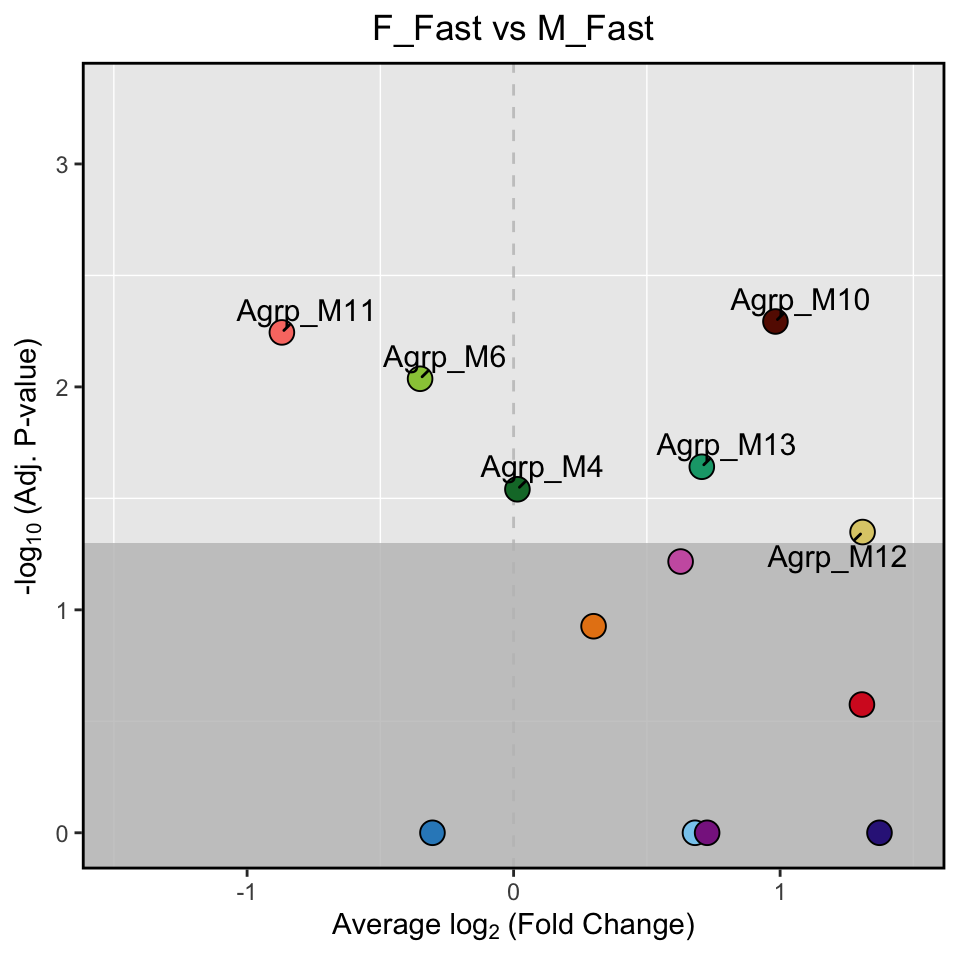
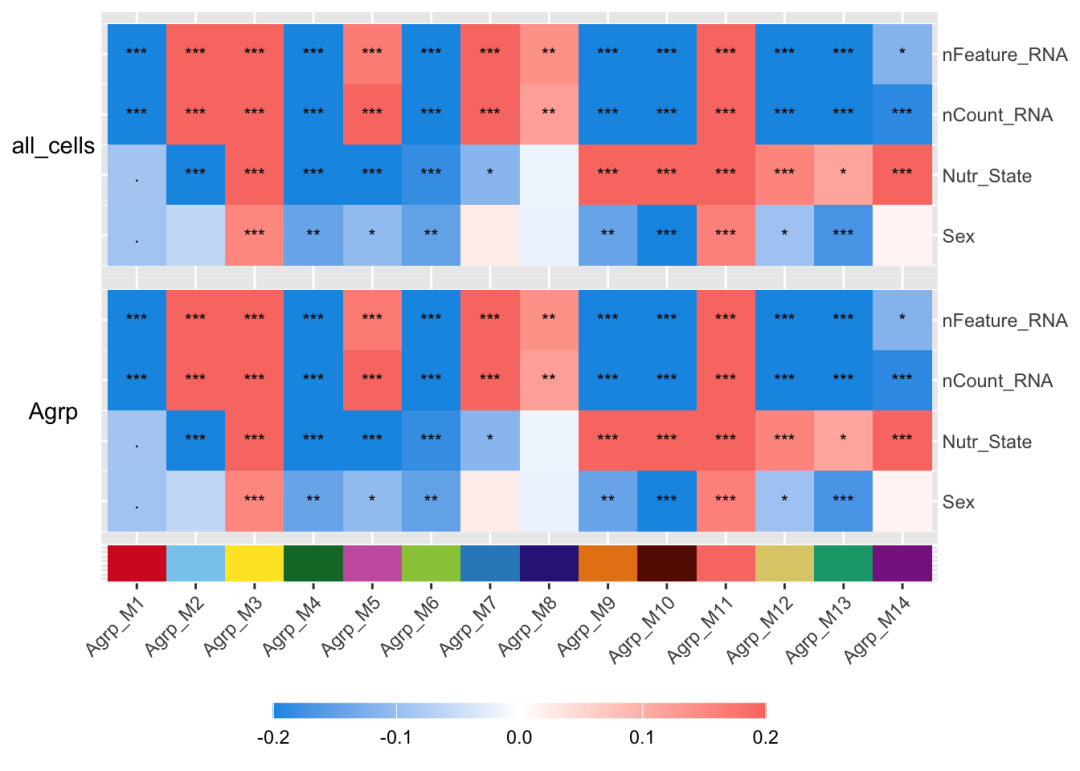
3模块性状相关性:Module Trait Correlation
这与WGCNA一致,将共表达模块与生物学变量和技术变量相关联,可以了解哪些模块与哪些性状(from seurat object metadata)关联,可以重点深入挖掘。使用ModuleTraitCorrelation函数,将选定的变量与模块特征基因进行相关性分析。该函数会计算特定细胞群组中的相关性,因为某些变量可能只在特定细胞群的某些模块中呈现相关性,而在其他细胞群中则不然。
#因为前面WGCNA是在agrp细胞上做的,所以这里做相关性将其提取子集
agrp_obj <- subset(seurat_obj, cell_type3 == "Agrp")
agrp_obj$Sex <- as.factor(agrp_obj$Sex)
agrp_obj$Sex <- factor(agrp_obj$Sex,levels = c("F","M"))
agrp_obj$Nutr_State <- as.factor(agrp_obj$Nutr_State)
agrp_obj$Nutr_State <- factor(agrp_obj$Nutr_State,levels = c("Fast","Fed"))
# list of traits to correlate
cur_traits <- c('Sex', 'Nutr_State','nCount_RNA', 'nFeature_RNA')
agrp_obj <- ModuleTraitCorrelation(
agrp_obj,
traits = cur_traits,
group.by='cell_type3',
wgcna_name = 'ARH'
)提取分析结果,这是一个list:包含相关性,p值,fdr三个文件。
Agrp_module_cor <- GetModuleTraitCorrelation(agrp_obj)
Agrp_module_cor可视化:
PlotModuleTraitCorrelation(
agrp_obj,
label = 'fdr',
label_symbol = 'stars',
text_size = 3,
text_digits = 3,
text_color = 'black',
high_color = '#F97B72',
mid_color = 'white',
low_color = '#1798E5',
plot_max = 0.2,
combine=TRUE
)
觉得我们分享有用的点个赞再走呗!
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-02,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号