ICLR 2026|35 个数据集、7 类任务:多肽 AI 迎来真正可比的 Benchmark

ICLR 2026|35 个数据集、7 类任务:多肽 AI 迎来真正可比的 Benchmark

MindDance
发布于 2026-06-01 18:17:45
发布于 2026-06-01 18:17:45

ICLR 2026 PepBenchmark: A Standardized Benchmark for Peptide Machine Learning
通用领域有刷不完的榜,肽这边连一张像样的统一考卷都凑不齐。肽被业内称作继小分子、单抗之后的第三代药物,合成方便、特异性高、安全性好,GLP-1类减肥降糖药这两年的火爆更是把它推到了聚光灯下。可一旦想用机器学习去做肽的活性、毒性、成药性预测,研究者面对的是一地散落的数据库、各家自定义的预处理、各跑各的评测指标。结果就是大家报出来的AUC一个比一个漂亮,却几乎没法横向比,谁也说不清进步到底是真本事还是刷出来的。中关村学院联合中国科学技术大学、南开大学、天津大学的团队,把这件脏活累活揽了下来,做出了 PepBenchmark。一句话:在严格切分下,蛋白语言模型(PLM)整体最强,分子指纹法在小数据和非天然肽上是硬通货,图神经网络和SMILES路线在单肽任务上反而拖后腿,而过去很多高分的真正元凶,是一个被忽视已久的问题。
一分钟速读:很火的大模型,遇上没人立规矩的肽数据
肽药研发的数据生态,用一团乱麻形容并不过分。抗菌肽、抗癌肽、神经肽、毒性肽散落在几十个数据库和论文附录里,每个研究者用之前都得自己淘洗一遍。淘洗的手法又五花八门:去冗余去不去、负样本怎么造、训练测试集怎么切,全凭各家习惯。隔壁小分子有 MoleculeNet 和 TDC,蛋白有 ProteinGym 和 TAPE,肽这边却始终没立起一个被广泛采用的基准。此前的三次尝试——UniDL4BioPep、Peptipedia、AutoPeptideML——要么数据规模有限,要么数据根本没做成可直接喂模型的形态,要么覆盖的任务太窄。
PepBenchmark 的野心就是把这三件事一次性焊死:统一的数据、统一的预处理流水线、统一的评测榜单。它由三块组成,PepBenchData 管数据,PepBenchPipeline 管处理,PepBenchLeaderboard 管评测。研究人员把这套东西打包成了一个同名 Python 包,几行代码就能加载预处理好的数据集、做标准切分、训练并评估。代码与数据已公开在 GitHub 仓库 ZGCI-AI4S-Pep/PepBenchmark。
https://github.com/ZGCI-AI4S-Pep/PepBenchmark/
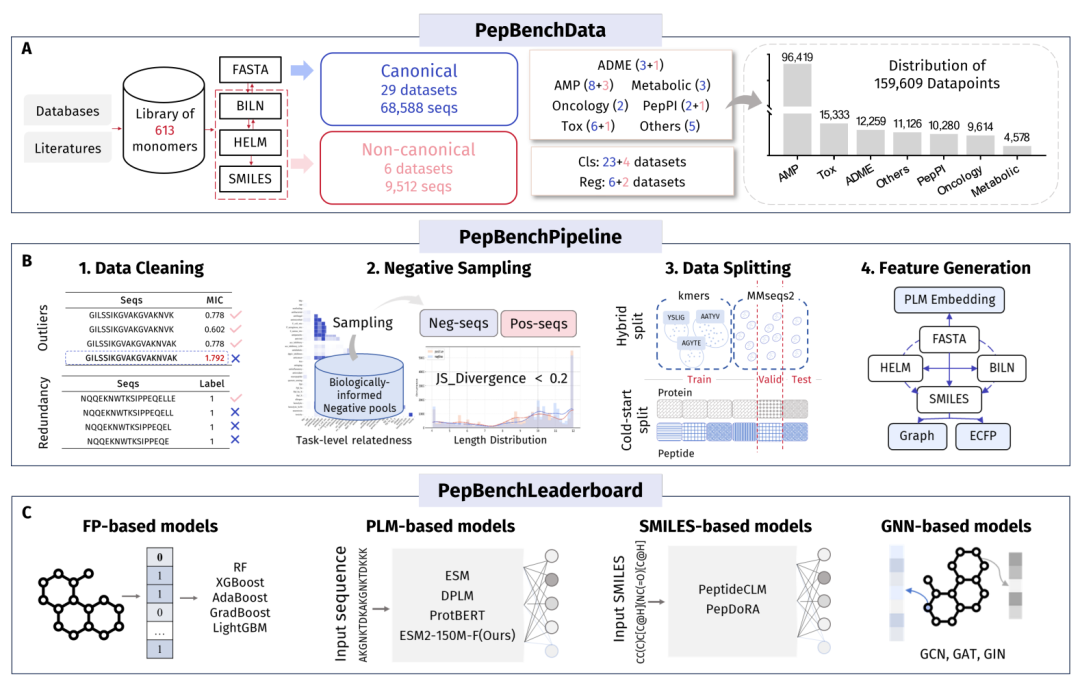
图 1 PepBenchmark 总体框架:数据、流水线、榜单三位一体
图 1 PepBenchmark 总体框架:数据、流水线、榜单三位一体
老毛病:为什么过去那些 90+ 的 AUC,很可能是刷出来的
这部分是全文最见功底的地方,团队没有急着秀模型,而是先把这个领域的顽疾一刀刀剖开。
第一刀是冗余。 肽药设计有个固有套路:拿一条已知活性的先导肽,做点突变、截短、延长,生成一大批近亲序列。这导致正样本里塞满了只差几个氨基酸的孪生兄弟。模型一旦见到这种数据,最省力的做法就是死记硬背那几个高频模式,而不是真去学序列到活性的映射。团队用 MMseqs2 在 90% 相似度阈值下做去重,统计出 9 个常用基准存在超过 5% 的冗余,其中溶血数据集冗余率高达 47%。去重之后,一个指纹法随机森林模型的 ROC-AUC 直接掉了 17.39%。这个数字相当扎心——它等于在说,之前那些漂亮分数里,有相当一部分是模型在背书,而不是在做题。
第二刀是负样本。 肽数据几乎全是正样本,负样本得人造,而怎么造一直没共识。常见两种做法都有坑:一种是从蛋白或数据库里随机切片当负样本,模型很容易学会区分活性肽和随机序列这种廉价差异,却没学到真正和目标性质相关的特征;另一种是拿别的活性肽当负样本,比如用抗菌肽给抗癌肽当反例,问题是这两类肽机制上高度重叠,很多抗癌肽本就是从抗菌数据里挖出来的,这么干等于往负样本里掺假阳性。更隐蔽的坑在于,正负样本在长度、电荷这些粗特征上分布一旦错开,模型直接抄这条捷径就能拿高分,评测结果自然虚高。
第三刀,也是最锋利的一刀,叫 kmer 泄漏。 团队发现,在多数肽数据集里,某些短片段(kmer)在正样本中出现的频率高得异常,这些往往是专家在设计肽时刻意引入的活性相关 motif。模型只要记住这些局部捷径就能拿分,完全不用理解背后的生物学。麻烦在于,业内常用的 MMseqs2 同源性切分根本管不了这个问题:两条序列可以共享同一个 kmer,整体相似度却很低,于是被分到了不同的数据集里,泄漏照样发生。团队拿一组模型在随机切分和 MMseqs2 切分下做对比,发现有些数据集(如 ace inhibitory、dppiv inhibitors)在 MMseqs2 切分下的分数竟然比随机切分还高——这个反直觉的现象,正是 kmer 泄漏在作祟。换句话说,光把序列拉开同源性远远不够,捷径还藏在更细的颗粒度里。
拆开看:怎么把一团乱麻,捋成一条标准流水线
诊断完了,PepBenchPipeline 给出的是一套四步走的硬约束流水线:数据清洗、负样本采样、数据切分、特征生成。这里头有两个真正硬核的设计。
第一个是负样本采样策略 BDNegSamp。 它定了两条铁律:正负样本的差异必须反映可泛化的生物学性质而非数据集特有的伪特征;同时尽量不引入假阴性。落地分三步走。先用所有收集到的活性肽搭一个生物学知情的负样本池,既避开随机序列的廉价差异,又保证负样本本身是真活性肽。接着做任务相关性过滤,这是最讲究的一环:团队用专家先验把肽分成三个高度相关的簇——膜相互作用类(抗菌、抗癌、溶血、跨膜)、糖代谢调节类(DPP-IV 抑制、抗糖尿病、抗氧化、抗炎)、神经活性类——再辅以序列重叠率统计(重叠率超过 0.05 即判定相关),把和目标任务机制相关的数据从负样本池里剔除,避免假阴性。最后是分布受控采样,在长度、净电荷、疏水性、氨基酸组成、二肽组成五个维度上,把正负样本的 Jensen-Shannon 散度压到极低——长度、电荷、疏水性约束在 0.2 以下,单氨基酸分布压到 0.05、二肽分布压到 0.15 以下。效果立竿见影:nonfouling 数据集的长度分布散度从 0.53 砍到 0.069,ttca 从 0.54 砍到 0.103,antimicrobial 从 0.35 砍到 0.021。这意味着模型再想靠长度、电荷这类捷径偷分,基本没门了。
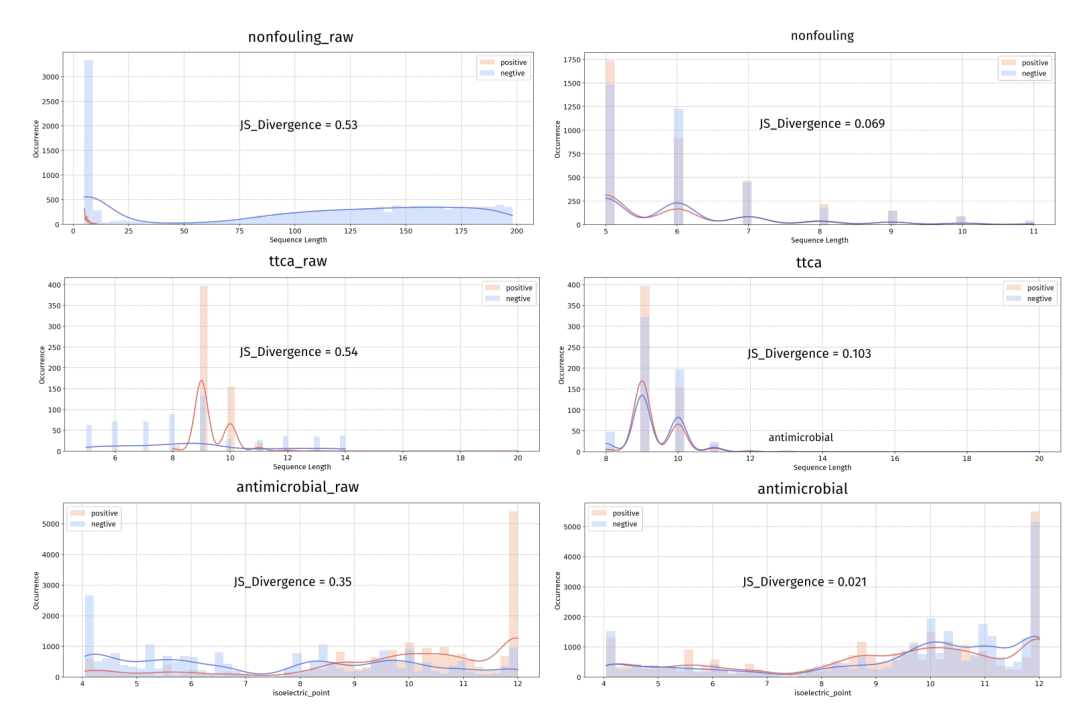
图 2 BDNegSamp 采样前后正负样本分布对比,散度大幅收敛意味着捷径特征被堵死
图 2 BDNegSamp 采样前后正负样本分布对比,散度大幅收敛意味着捷径特征被堵死
第二个硬核设计是切分策略。 针对 kmer 泄漏,团队先用 Fisher 精确检验(配合 Benjamini-Hochberg 做 FDR 校正)找出在正样本里显著富集的 motif,然后把共享同一 motif 的序列归到同一个簇,按 8:1:1 整簇分配到训练、验证、测试集——这样任何一个富集 motif 都不会跨集出现,逼着模型去做真正的跨 motif 泛化。在此基础上叠加 MMseqs2 同源性聚类(身份阈值定在 0.3,此时序列聚得最充分),就得到了团队主推的 hybrid-split。它同时堵住 kmer 泄漏和全局同源,是一套明显更严苛、也更贴近真实成药场景的评测协议。对于非天然肽,序列工具搞不定,就改用 ECFP 指纹算 Tanimoto 相似度建图,把连通分量当簇来切;蛋白-肽互作(PepPI)任务则用 cold-start,保证蛋白不跨集。
还有一个巧思值得点出来。 非天然肽序列太少,凑不出像样的负样本池,团队干脆训了一个 GPT-2 的序列到序列生成模型,把天然肽翻译成带化学修饰的非天然肽(BILN 格式),等于把非天然肽的负采样难题,转化回了天然肽空间里去解。这个生成器的 BILN 转 SMILES 成功率 89.7%,化学有效性 100%,新颖性比例 76.5%,工程上相当扎实。
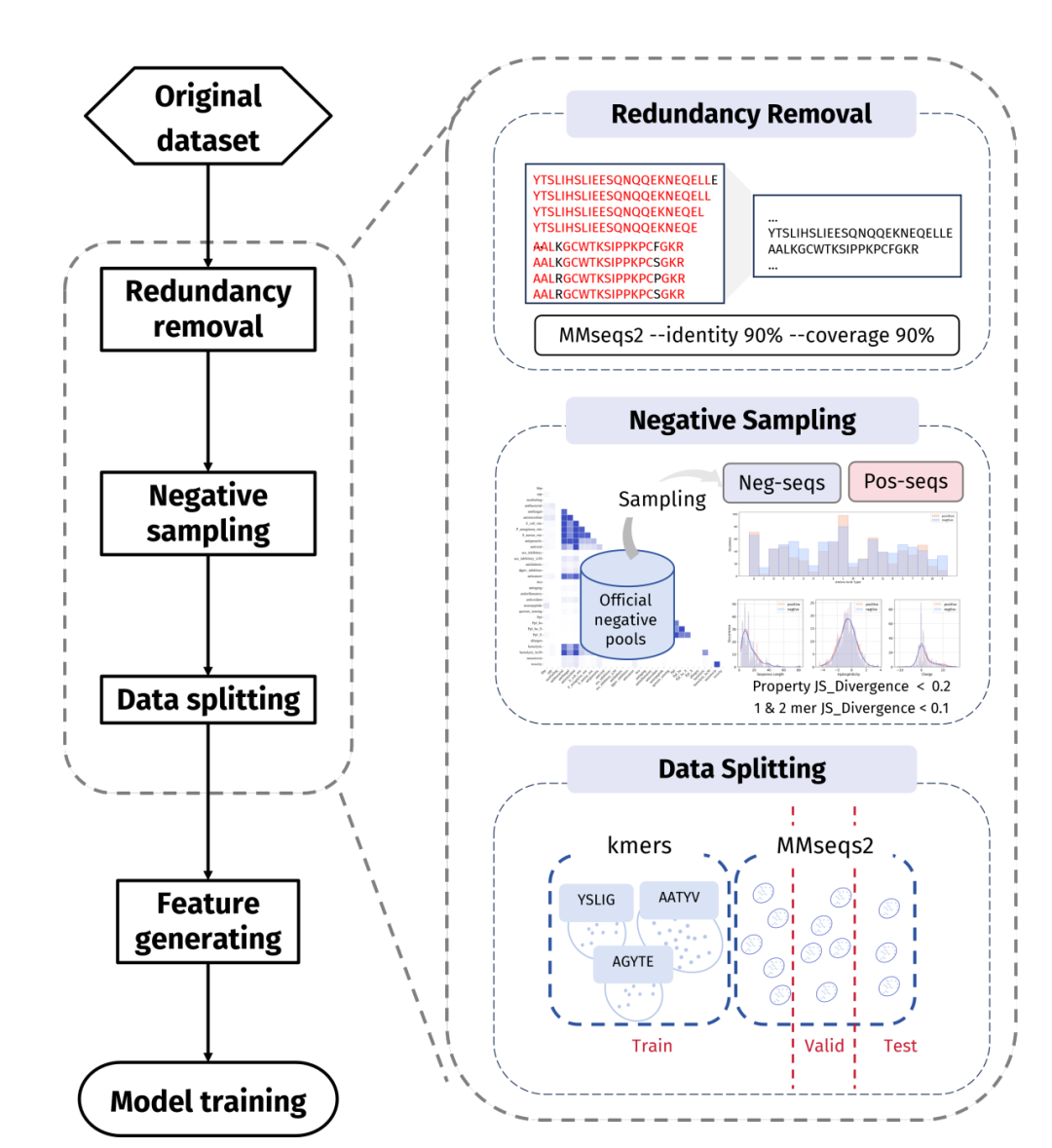
图 3 PepBenchPipeline 数据集构建流水线全景
图 3 PepBenchPipeline 数据集构建流水线全景
跑分见真章:到底谁强谁弱,数据说了算
评测覆盖四大模型家族:指纹法(RF、XGBoost、LightGBM 等基于 1024 位 ECFP6)、图神经网络(GCN、GAT、GIN,以及肽预训练的 Pepland)、SMILES 法(ChemBERTa、PepDoRA、PeptideCLM)、蛋白语言模型(ESM2 全家族、DPLM、ProtBERT)。团队还把 ESM2-150M 在 193 万条短肽上继续预训练,做出了自家的 ESM-150M-F。在 hybrid-split 这把严尺子下,单肽分类任务的平均 ROC-AUC 排出了清晰的座次:ESM-150M-F 以 81.5 拿下 SOTA,DPLM-150M 80.9 紧随其后,ESM2 各档位在 80 上下,随机森林 79.2 排在第三,SMILES 的 ChemBERTa 73.2、PepDoRA 62.1,图神经网络的 GIN 69.5、Pepland 63.9 垫底。这张表信息量极大,几个判断可以直接落地。
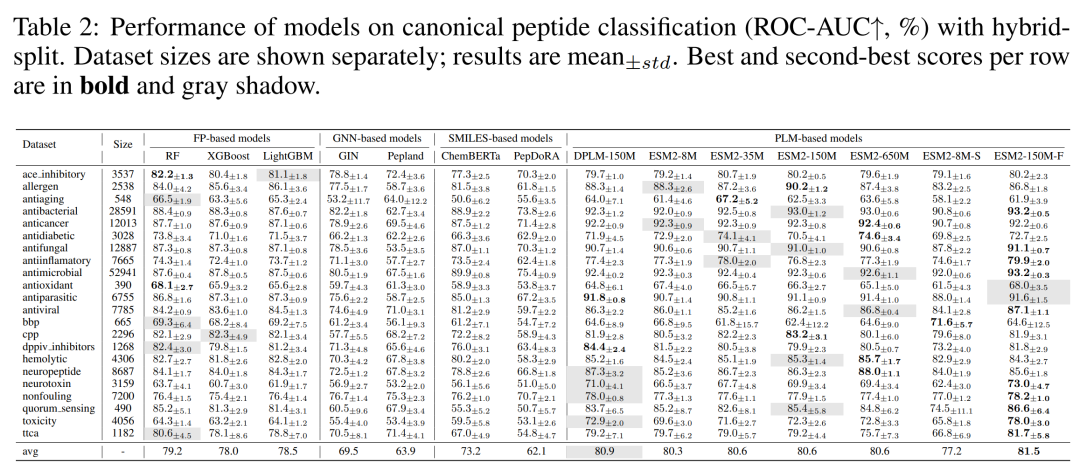
天然肽分类任务各模型表现:PLM 居首,指纹法第二,图/SMILES 路线落后
天然肽分类任务各模型表现:PLM 居首,指纹法第二,图/SMILES 路线落后
第一个判断,表征方式决定上限。 对只由 20 种标准氨基酸组成的天然肽来说,原子级表征(图、SMILES)引入的是冗余而非信息,序列级的 PLM 反而把蛋白预训练的知识迁移了过来。第二个判断更出人意料:预训练并非总是好事。 从随机初始化的 ESM2-8M-S(77.2)到预训练的 ESM2-8M(80.3),大规模蛋白预训练的收益清清楚楚;可 Pepland 和 PepDoRA 这两个专门在肽数据上做图/SMILES 预训练的模型,表现竟然不如没预训练的 GIN 和 ChemBERTa——这是典型的负迁移,说明在不合适的表征上硬做领域预训练会帮倒忙,怎么破解还是个开放问题。第三个判断,指纹法是小数据的定海神针。 RF 配 ECFP6 在 ace inhibitory、antiaging、antioxidant、bbp、dppiv 这些小数据集上频频拿第一第二,而 ECFP6 本是为小分子设计的,在肽上还能打成这样,反过来说明专为肽量身定做的指纹是一个很有前景的方向。
非天然肽这边的结论更反直觉。按常理,600 多种非天然单体带来的化学多样性,应该让原子级表征如鱼得水,可实测下来,指纹法(RF/XGBoost 平均接近 96)依旧吊打图和 SMILES。这说明现有的图/SMILES 路线还远没把非天然肽的模块化结构学明白。而到了蛋白-肽互作任务,剧情又翻转了:图和 SMILES 在这里变得有竞争力,反衬出原子级的精细分子细节,对建模分子间相互作用比建模单肽性质更管用。一个有意思的细节是,在更大的 ppi 数据集上,PLM 越大反而越差,出现了性能退化——这提醒大家,scaling law 在跨分子互作任务上未必照搬有效。
自家的 ESM-150M-F 也讲了个干净的故事。ESM2 在 UniRef 上预训练,但 50 残基以下的短肽只占 UniRef50 的 2.8%,严重欠拟合。团队把 UniRef50 截到 50 残基以下,凑出 193 万条短肽继续预训练,短肽上的伪困惑度明显下降。这一刀补在了分类任务上效果显著,回归任务提升却有限,说明回归更受母模型本身容量的约束。
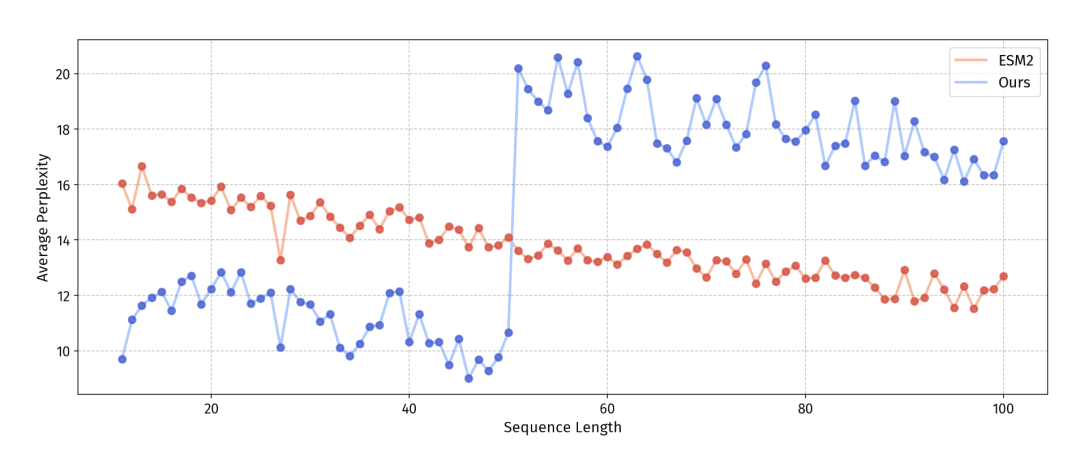
图 4 短肽微调让 ESM-150M-F 在短序列上的困惑度全面低于原版 ESM2
图 4 短肽微调让 ESM-150M-F 在短序列上的困惑度全面低于原版 ESM2
余味:肽药 AI 的下一步,可能要去敲 HPC 的门
PepBenchmark 真正的价值,不在于又造了个榜,而在于它把这个领域的评测地基重新浇了一遍。当大家都用同一套数据、同一条流水线、同一把尺子,所谓的方法进步才第一次变得可被检验。那些被冗余和 kmer 泄漏吹起来的虚高分数,在 hybrid-split 面前会现出原形——这对一个想从论文走向临床的领域,是迟早要补上的一课。
往前看,团队自己点出了最大的短板:缺结构数据。整个 PDB 里只有约 2000 条肽,其中非天然肽不到 100 条,基于结构的预测和生成任务暂时无从评测。他们给出的解法很值得玩味:既然实验结构太稀缺,那就靠计算模拟补。肽分子小,通常只有几百个原子,这恰恰是 QM/MM 混合方法、增强分子动力学这类高精度模拟的舒适区。换句话说,肽药 AI 的下一程,很可能要和 HPC、大规模物理模拟深度绑定——用算力换数据,再用数据喂模型,形成一个闭环。这条路一旦走通,AIDD 的范式或许会从今天的纯数据驱动,慢慢挪向数据与第一性原理模拟并跑的混合模式。
立好了尺子,接下来该量的东西还很多。专为肽设计的指纹、能正确做领域预训练而不负迁移的框架、面向非天然肽模块化结构的新表征、以及结构层面的标准任务——每一个都是留给后来者的开放命题。基准的意义,从来不是终点,而是让所有人站到同一条起跑线上。
参考文献
Jiahui Zhang, Rouyi Wang, Kuangqi Zhou, Tianshu Xiao, Lingyan Zhu, Yaosen Min, Yang Wang. PepBenchmark: A Standardized Benchmark for Peptide Machine Learning. ICLR 2026. 代码与数据: https://github.com/ZGCI-AI4S-Pep/PepBenchmark/
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号