Esmfold2食用体验-蛋白质结构预测

Esmfold2食用体验-蛋白质结构预测

Tom2Code
发布于 2026-06-01 15:35:31
发布于 2026-06-01 15:35:31
这里引用一下biohub的官网的一句话:
Biohub releases a world model of
protein biology
所以,这就是biohub如何介绍的esmfold2,在引入一段介绍:
- ESMFold2 is the design engine built to transform ESMC’s sequence representations into atomically-resolved 3D structure of biomolecular complexes. In experiments described in a preprint posted today, researchers used ESMFold2 to design protein binders against five targets central to cancer and immunology — a computational search completed in days, rather than several months or years. The lab-validated binders exhibited high affinity, specificity, and stability — properties critical for clinical utility — and showed minimal similarity to sequences in public databases, suggesting the model is producing de novo solutions, rather than retrieving known binders.
原文翻译:
ESMFold2是一款设计引擎,旨在将ESMC的序列表示转化为生物分子复合物的原子级分辨三维结构。在今天发布的一篇预印本中描述的实验中,研究人员使用ESMFold2设计了针对癌症和免疫学领域五个核心靶点的蛋白质结合剂——这一计算搜索仅在几天内完成,而非几个月或几年。经过实验室验证的结合剂表现出高亲和力、特异性和稳定性——这些特性对于临床应用至关重要——并且与公共数据库中的序列相似度极低,表明该模型正在生成全新的解决方案,而非检索已知的结合剂。
虽然官网说esmfold2可以设计蛋白质,但是官网给的代码的demo都是蛋白质结构的预测:

官方教程展示了 ESMFold2 的三个代表性应用场景。
第一类是蛋白质-核酸复合物预测,例如 LC11-RNase H1 与 RNA 杂合双链形成的复合体系,这说明 ESMFold2 能够同时处理蛋白质链、RNA 链和 DNA 链,并对它们之间的空间关系进行建模。
第二类是含有非标准氨基酸和共价修饰的体系,例如 GLP-1 受体与司美格鲁肽的复合物。司美格鲁肽是一类治疗性多肽,包含非标准氨基酸以及共价连接的脂质 linker。该示例说明 ESMFold2 可以通过 CCD 代码定义非天然残基,通过 SMILES 描述小分子配体,并显式指定多肽和配体之间的共价键,从而处理更接近真实药物分子的复杂结构。
此外,教程的第三类还介绍了如何在结构预测中引入 MSA,即多序列比对信息。MSA 能够提供蛋白质家族中的进化保守性和共进化关系,这些信息有助于模型判断哪些残基之间可能存在空间接触,从而提高结构预测的准确性。教程以泛素为例,说明在已有进化信息的情况下,ESMFold2 可以进一步利用 MSA 改善预测效果。总体来看,这一教程想表达的是:ESMFold2 不只是一个普通的蛋白质折叠工具,而是一个能够处理蛋白质、核酸、小分子、非标准残基、共价修饰以及多组分复合物的综合性结构预测框架。
读者大佬们要是想使用esmfold2进行结构预测,请自行去esmfold2申请api key:
https://biohub.org/
接下来就是食用教程了
01.依赖安装
本教程是tom在colab中运行的,所以需要先执行下面的这段:
# If you are working in colab, uncomment these lines to install dependencies
!pip install esm@git+https://github.com/Biohub/esm.git@c94ed8d
!pip install py3dmol
然后是依赖
from getpass import getpass
import matplotlib.patches as mpatches
import matplotlib.pyplot as plt
import numpy as np
import py3Dmol
from esm.sdk import esmfold2_client
from esm.sdk.api import FoldingConfig
from esm.utils.structure import input_builder
定义画图的函数:
from IPython.display import HTML, display
def plddt_hex(v):
"""Convert pLDDT score to hex color."""
if v >= 90:
return"#0053D6"
if v >= 70:
return"#65CBF3"
if v >= 50:
return"#FFDB13"
return"#FF7D45"
PLDDT_LEGEND = (
'<span style="color:#FF7D45;">■</span> <50 '
'<span style="color:#FFDB13;">■</span> 50–70 '
'<span style="color:#65CBF3;">■</span> 70–90 '
'<span style="color:#0053D6;">■</span> >90'
)
def normalize_plddt(plddt):
plddt = np.array(plddt)
return plddt * 100if plddt.max() <= 1.0else plddt
def apply_plddt_style(view, plddt, add_hetflag=False):
view.setStyle({}, {})
for i, s in enumerate(plddt):
view.setStyle({"resi": i + 1}, {"cartoon": {"color": plddt_hex(s)}})
if add_hetflag:
view.addStyle({"hetflag": True}, {"stick": {}})
view.zoomTo()
def show_dual_viewer(v1, title1, legend1, v2, title2, legend2):
l1 = (
f'<p style="text-align:center; font-size:12px; margin-top:4px;">{legend1}</p>'
if legend1
else""
)
l2 = (
f'<p style="text-align:center; font-size:12px; margin-top:4px;">{legend2}</p>'
if legend2
else""
)
display(
HTML(
'<div style="display:flex; gap:20px;">'
f'<div><p style="text-align:center; font-weight:bold;">{title1}</p>'
+ v1._make_html()
+ l1
+ "</div>"
f'<div><p style="text-align:center; font-weight:bold;">{title2}</p>'
+ v2._make_html()
+ l2
+ "</div>"
"</div>"
)
)
def plot_pae(pae, chains=None, title=None):
"""Plot a PAE matrix with optional per-chain colored bars and legend.
Args:
pae: PAE as a numpy array (n x n).
chains: list of (start, end, label, color) tuples. When provided, colored
bars are drawn along the top and left axes and a legend is shown.
title: optional plot title.
"""
total_len = pae.shape[0]
fig, ax = plt.subplots(figsize=(8, 8))
im = ax.imshow(
pae, cmap="Greens_r", vmin=0, vmax=30, origin="upper", aspect="equal"
)
if chains:
bar_thickness = total_len * 0.015
for start, end, label, color in chains:
ax.add_patch(
mpatches.Rectangle(
(start - 0.5, -bar_thickness - 1),
end - start,
bar_thickness,
facecolor=color,
clip_on=False,
edgecolor="black",
linewidth=0.5,
)
)
ax.add_patch(
mpatches.Rectangle(
(-bar_thickness - 1, start - 0.5),
bar_thickness,
end - start,
facecolor=color,
clip_on=False,
edgecolor="black",
linewidth=0.5,
)
)
for start, *_ in chains[1:]:
ax.axhline(start - 0.5, color="black", linewidth=1.5)
ax.axvline(start - 0.5, color="black", linewidth=1.5)
legend_handles = [
mpatches.Patch(facecolor=c, edgecolor="black", label=l)
for _, _, l, c in chains
]
ax.legend(
handles=legend_handles,
loc="upper center",
bbox_to_anchor=(0.5, -0.08),
ncol=len(chains),
frameon=True,
fontsize=10,
title="Chain",
)
ax.set_xlim(-bar_thickness - 2, total_len)
ax.set_ylim(total_len, -bar_thickness - 2)
ax.set_xlabel("Scored Residue", fontsize=12)
ax.set_ylabel("Aligned Residue", fontsize=12)
if title:
ax.set_title(title, fontsize=14, fontweight="bold", pad=15)
cbar = fig.colorbar(im, ax=ax, fraction=0.046, pad=0.04)
cbar.set_label("Expected Position Error (Ångströms)", fontsize=12)
cbar.set_ticks([0, 5, 10, 15, 20, 25, 30])
cbar.ax.invert_yaxis()
plt.tight_layout()
plt.show()
02输入api key
首先,我们需要在 Biohub 平台上生成一个 API 密钥 (API Key),并将其配置到你的账户中。这个密钥是用来管理你的调用额度 (credits) 和访问权限的。
token = getpass("Biohub token: ")
client = esmfold2_client(
model="esmfold2-fast-2026-05", url="https://biohub.ai", token=token
)
03folding预测参数设置
在进行结构预测时,可以通过设置折叠参数来控制预测质量与计算成本之间的平衡。常用参数主要包括 num_loops 和 num_sampling_steps。
其中,num_loops 表示模型对结构进行迭代优化的次数。一般来说,迭代次数越多,预测结构可能越精细、准确性也可能越高,但相应的计算时间也会增加。通常情况下,该参数可以设置在 3–5 之间。
num_sampling_steps 表示扩散采样的步数。采样步数越多,模型生成结构时的搜索过程越充分,预测结果通常也会更加准确,但运行时间也会明显延长。一般推荐范围为 200–400。
在本示例中,我们采用了相对适中的参数设置:num_loops 设置为 3,num_sampling_steps 设置为 32,以便在预测精度和运行效率之间取得平衡。对于需要更高精度的预测任务,尤其是在模拟柔性配体或结合模式不确定的复合物时,可以适当增加 num_loops 和 num_sampling_steps,但这也会带来更高的计算成本和更长的运行时间。
config = FoldingConfig(num_loops=3, num_sampling_steps=32, include_pae=True)
04第一个案例
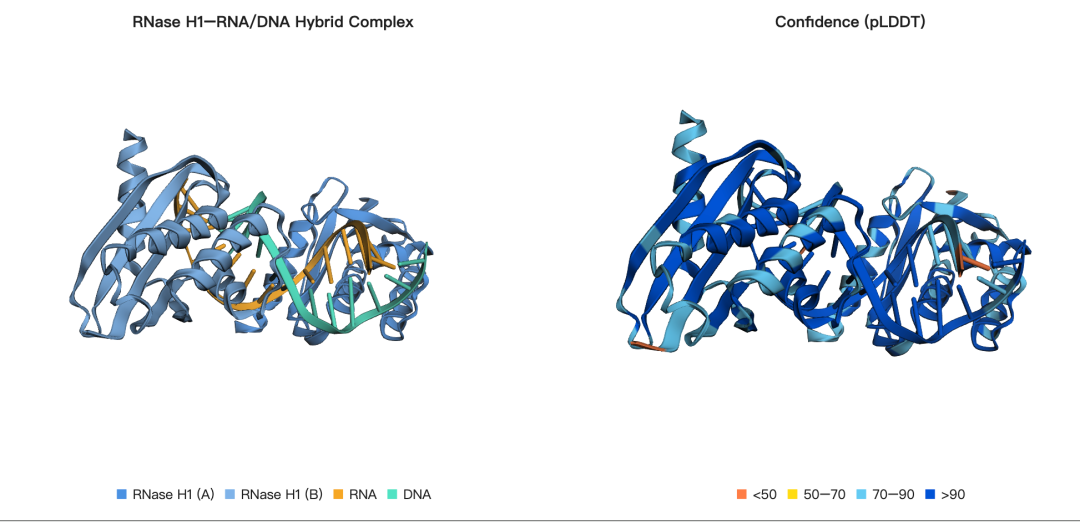
示例 1:RNase H1–RNA/DNA 杂合复合物
核糖核酸酶 H1(Ribonuclease H1,RNase H1)是一类内切核酸酶,能够特异性水解 RNA:DNA 杂合双链中的 RNA 链。RNase H 活性在 DNA 复制、转录以及逆转录过程中都具有重要作用,同时也是 gapmer 类反义寡核苷酸药物发挥作用的重要机制。
本示例选取的复合物来自 PDB 结构 4H8K。该体系由四条链组成,并包含三类不同的生物学组分:
LC11-RNase H1(链 A 和链 B):这是一种来源于宏基因组的热稳定 RNase H1 同源蛋白,在实验中以同源二聚体形式表达并结晶。在建模时,我们将其作为一个 ProteinInput 输入,并指定两个链 ID。
底物 RNA(链 C):该 RNA 链与 DNA 模板链配对形成 RNA:DNA 杂合双链,并作为酶切反应中的底物,被 RNase H1 识别和切割。
底物 DNA(链 D):该 DNA 链是杂合双链中的互补链,与 RNA 链共同构成 RNA:DNA hybrid duplex。
在这一示例中,我们将同时对这四条链进行结构折叠预测,以尽可能重现 PDB 4H8K 中观察到的复合物空间构象与整体架构。该案例展示了 ESMFold2 在处理蛋白质-核酸复合物方面的能力,尤其适用于同时包含蛋白质、RNA 和 DNA 多组分体系的结构建模。
# Define all components from PDB 4H8K (LC11-RNase H1 + RNA/DNA hybrid)
# LC11-RNase H1 protein sequence (140 amino acids, chains A and B share this sequence)
rnaseh_sequence = (
"MNKIIIYTDGGARGNPGPAGIGVVITDEKGNTLHESSAYIGETTNNVAEYEALIRALEDLQ"
"MFGDKLVDMEVEVRMNSELIVRQMQGVYKVKEPTLKEKFAKIAHIKMERVPNLVFVHIPRE"
"KNARADELVNEAIDKALS"
)
# Substrate RNA sequence (14 nucleotides)
rna_sequence = "CGACACCUGAUUCC"
# Substrate DNA sequence (14 nucleotides, complementary to the RNA)
dna_sequence = "GGAATCAGGTGTCG"
# Express the homodimer as a single ProteinInput with two chain IDs
rnaseh_protein = input_builder.ProteinInput(id=["A", "B"], sequence=rnaseh_sequence)
substrate_rna = input_builder.RNAInput(id="C", sequence=rna_sequence)
substrate_dna = input_builder.DNAInput(id="D", sequence=dna_sequence)
# Create the complex input
rnaseh_complex_input = input_builder.StructurePredictionInput(
sequences=[rnaseh_protein, substrate_rna, substrate_dna]
)
rnaseh_structure = client.fold_all_atom(rnaseh_complex_input, config=config)
# Save the structure
with open("rnaseh_complex_4h8k.cif", "w") as f:
f.write(rnaseh_structure.complex.to_mmcif())
可视化:
rnaseh_cif = open("rnaseh_complex_4h8k.cif").read()
rnaseh_plddt = normalize_plddt(rnaseh_structure.plddt.cpu().numpy())
v1 = py3Dmol.view(width=500, height=400)
v1.addModel(rnaseh_cif, "mmcif")
v1.setStyle({"chain": "A"}, {"cartoon": {"color": "#4A90E2"}})
v1.setStyle({"chain": "B"}, {"cartoon": {"color": "#7FB3E8"}})
v1.setStyle({"chain": "C"}, {"cartoon": {"color": "#F5A623"}})
v1.setStyle({"chain": "D"}, {"cartoon": {"color": "#50E3C2"}})
v1.zoomTo()
v2 = py3Dmol.view(width=500, height=400)
v2.addModel(rnaseh_cif, "mmcif")
apply_plddt_style(v2, rnaseh_plddt)
rnaseh_legend = (
'<span style="color:#4A90E2;">■</span> RNase H1 (A) '
'<span style="color:#7FB3E8;">■</span> RNase H1 (B) '
'<span style="color:#F5A623;">■</span> RNA '
'<span style="color:#50E3C2;">■</span> DNA'
)
show_dual_viewer(
v1,
"RNase H1–RNA/DNA Hybrid Complex",
rnaseh_legend,
v2,
"Confidence (pLDDT)",
PLDDT_LEGEND,
)
输出:
05第二个案例
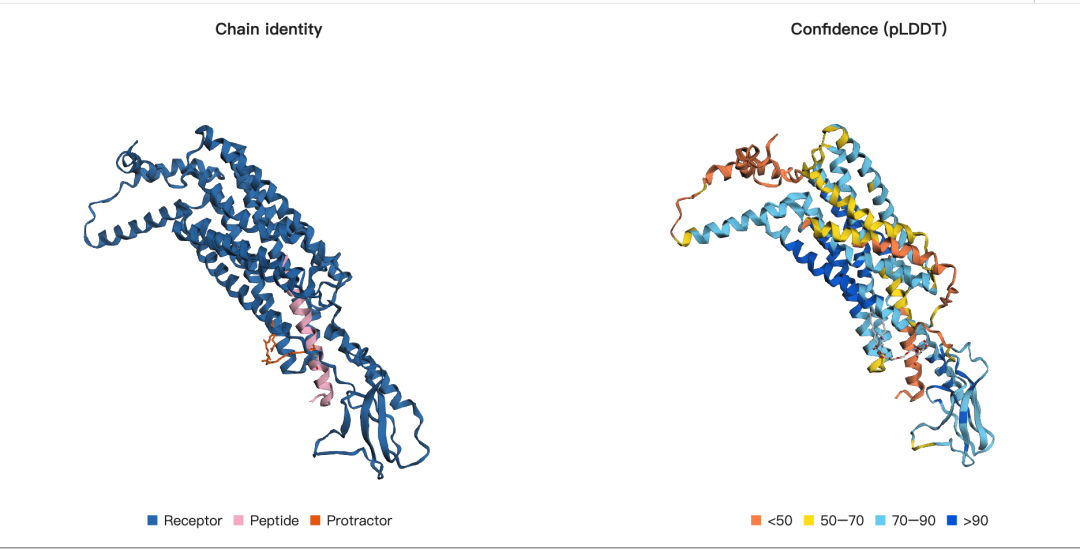
示例 2:司美格鲁肽与 GLP-1 受体的复合物结构预测
这段代码定义的是人源 GLP-1 受体(GLP1R)的蛋白质序列,序列来源对应 UniProt 编号 P43220。GLP-1 受体是一种典型的跨膜 G 蛋白偶联受体(GPCR),也是司美格鲁肽等 GLP-1 类药物的主要作用靶点。在本示例中,模型需要预测的是司美格鲁肽与 GLP-1 受体结合形成的复合物结构,因此首先需要将受体蛋白作为一个输入对象提供给模型。
需要注意的是,这里输入的并不是单纯的天然 GLP-1R 序列,而是一个实验构建体序列。该构建体包含 N 端的 FLAG 标签、TEV 蛋白酶切割位点,以及 C 端的 His 标签。FLAG 标签和 His 标签常用于蛋白表达、纯化和检测;TEV cleavage site 则用于在实验中通过 TEV 蛋白酶切除标签区域。因此,这条序列更接近实验结构解析或重组蛋白表达中使用的受体构建体,而不是完全未修饰的天然蛋白序列。
# Human GLP1R sequence (UniProt P43220)
# This construct includes N-terminal FLAG tag, TEV cleavage site, and C-terminal His-tag
receptor_sequence = (
"MKTIIALSYIFCLVFADYKDDDDLEVLFQGPARPQGATVSLWETVQKWREYRRQCQRSLTEDPPPATDLFCNRTFDEYAC"
"WPDGEPGSFVNVSCPWYLPWASSVPQGHVYRFCTAEGLWLQKDNSSLPWRDLSECEESKRGERSSPEEQLLFLYIIYTVG"
"YALSFSALVIASAILLGFRHLHCTRNYIHLNLFASFILRALSVFIKDAALKWMYSTAAQQHQWDGLLSYQDSLSCRLVFL"
"LMQYCVAANYYWLLVEGVYLYTLLAFSVFSEQWIFRLYVSIGWGVPLLFVVPWGIVKYLYEDEGCWTRNSNMNYWLIIRL"
"PILFAIGVNFLIFVRVICIVVSKLKANLMCKTDIKCRLAKSTLTLIPLLGTHEVIFAFVMDEHARGTLRFIKLFTELSFT"
"SFQGLMVAILYCFVNNEVQLEFRKSWERWRLEHLHIQRDSSMKPLKCPTSSLSSGATAGSSMYTATCQASCSPAGLEVLF"
"QGPHHHHHHH"
)
receptor = input_builder.ProteinInput(id="A", sequence=receptor_sequence)
定义司美格鲁肽多肽链
接下来,我们需要定义司美格鲁肽多肽链,在该示例中将其设定为 B 链。与普通天然多肽不同,司美格鲁肽是一种经过人工修饰的治疗性多肽,因此在建模时需要显式指定其中的非标准氨基酸和后续脂质 linker 的连接位点。
首先,司美格鲁肽在第 2 位引入了 AIB 替换。AIB 的全称是 α-aminoisobutyric acid,α-氨基异丁酸,是一种非天然氨基酸。它的作用是限制多肽主链的柔性,从而提高多肽整体构象稳定性,并降低其被蛋白酶降解的可能性。这类修饰在治疗性多肽药物中非常常见,主要目的是改善药物的体内稳定性和药代动力学性质,例如延长半衰期、提高抗降解能力等。
其次,需要确定多肽中的 赖氨酸残基 作为脂质 linker 的连接位点。在司美格鲁肽中,特定赖氨酸侧链会与脂质化修饰基团形成共价连接。这个脂质 linker 对药物性质非常关键,它可以增强司美格鲁肽与血清白蛋白的结合能力,从而延长药物在体内的循环时间。因此,在全原子结构预测中,除了定义多肽本身,还需要明确指出哪个赖氨酸残基将与脂质 linker 形成共价键,以便模型能够正确构建完整的修饰多肽结构。
# Semaglutide peptide sequence
peptide_sequence = "HAEGTFTSDVSSYLEGQAAKEFIAWLVRGRG"
# AIB substitution (0-indexed)
aib_position = 1
peptide = input_builder.ProteinInput(
id="B",
sequence=peptide_sequence,
modifications=[input_builder.Modification(position=aib_position, ccd="AIB")],
)
lys_idx = peptide_sequence.index("K") # residue index in peptide (0-based)
print("Lys at residue index:", lys_idx)
输出:

next:
定义脂质 Linker(SMILES)
接下来,需要使用 SMILES 表示法定义一个类似脂质的 linker 结构。SMILES 是一种用字符串描述化学分子结构的常用方式,可以明确表示分子中的原子连接关系、键类型以及部分结构特征。
在这个示例中,脂质 linker 的 SMILES 被特意写成一种便于建模的形式:将用于连接多肽赖氨酸侧链的 羰基碳原子 放在 SMILES 字符串的第一个原子位置。这样做的好处是可以简化后续的原子编号和共价键指定过程。也就是说,当我们需要告诉模型“脂质 linker 的哪个原子要与赖氨酸形成共价键”时,由于连接位点已经被设置为第一个原子,因此更容易准确指定该原子,避免复杂分子中原子索引混乱的问题。
这一部分的核心目的,是为司美格鲁肽中的脂质化修饰提供一个明确的小分子结构输入。通过 SMILES 定义 linker,并进一步指定其与多肽赖氨酸残基之间的共价连接关系,模型才能在后续全原子结构预测中正确构建完整的修饰多肽结构。
def get_protractor_smiles() -> str:
# "Protractor" refers to Novo Nordisk's fatty acid linker technology used in semaglutide.
# This SMILES is a representative structure inspired by semaglutide's linker design,
# not the exact compound. For the precise semaglutide structure, see PDB 7KI0 or the FDA label.
# First atom (index 0) is the carbonyl carbon used for attachment to the peptide.
return "C(=O)(CCOCCOCC(=O)NCCOCCOCCNCC(=O)N[C@@H](CCC(=O)NCCCCCCCCCCCCCCCCCC(=O)O)C(=O)O)"
ligand = input_builder.LigandInput(id="C", ccd=None, smiles=get_protractor_smiles())
next:
引入共价键并折叠复合物
在完成受体蛋白、司美格鲁肽多肽链以及脂质 linker 的定义之后,接下来就可以将三者作为一个整体进行结构预测。也就是说,模型需要同时折叠 人源 GLP-1 受体、司美格鲁肽多肽以及脂质化 linker,并在建模过程中显式考虑多肽与 linker 之间形成的共价键。
在指定共价键时,一个关键问题是需要准确识别参与连接的具体原子。对于司美格鲁肽这一侧,共价连接发生在 B 链中的赖氨酸残基上。赖氨酸侧链末端的氨基原子通常记为 NZ,它是形成脂质化修饰共价连接的重要反应位点。
在该示例中,代码使用 atom_idx1=8 来指定赖氨酸残基中的连接原子。这是因为在常见的 0-based 编号体系中,赖氨酸残基的 NZ 原子通常是第 9 个原子,对应索引值为 8。需要注意的是,不同输入格式或不同建模工具中,原子编号方式可能并不完全一致,因此在实际使用时,应根据完整结构或残基定义文件确认该原子索引是否正确。
对于任意一个残基,其内部原子顺序通常可以参考对应的化学组分定义文件。例如,赖氨酸的原子排序可以在 Lysine 的 CIF 文件中通过 pdbx_ordinal 字段查看。这个字段记录了残基内部各原子的标准排序,因此可以帮助我们准确确定 NZ 等关键原子的索引位置。通过这种方式,模型才能明确知道“多肽中的哪个原子”需要与“脂质 linker 中的哪个原子”形成共价键,从而正确构建完整的司美格鲁肽脂质化修饰结构。
bond = input_builder.CovalentBond(
chain_id1="B", res_idx1=lys_idx, atom_idx1=8, chain_id2="C", res_idx2=0, atom_idx2=0
)
complex_inputs = input_builder.StructurePredictionInput(
sequences=[receptor, peptide, ligand], covalent_bonds=[bond]
)
complex_structure = client.fold_all_atom(complex_inputs, config=config)
with open("glp1r_semaglutide.cif", "w") as f:
f.write(complex_structure.complex.to_mmcif())
最后就是结构的可视化了:
06第三个例子:
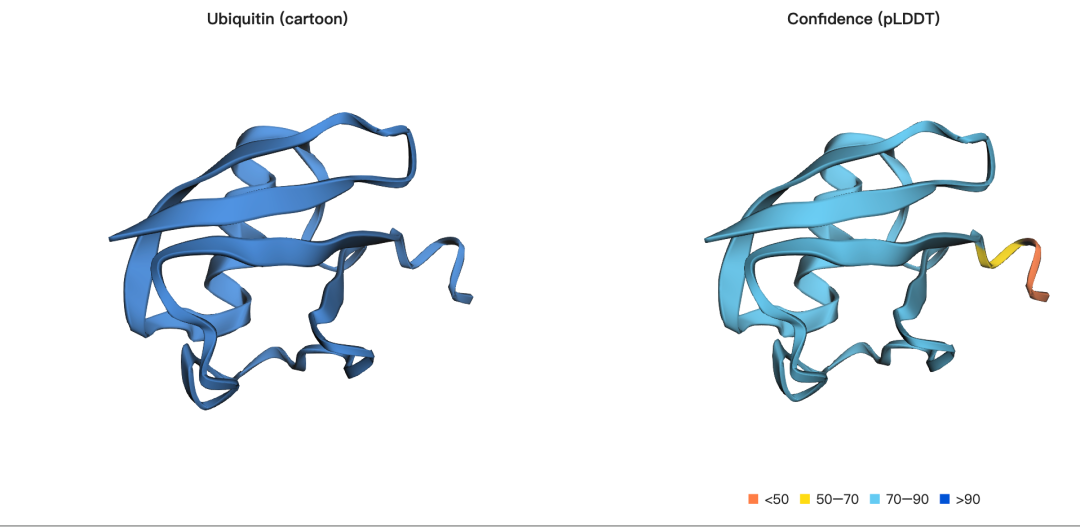
示例 3:基于 MSA 引导的泛素结构折叠
在前面的示例中,我们主要使用的是单序列结构预测方式。单序列折叠的优势是速度较快、输入简单,但在某些情况下会牺牲一定的预测精度。除了单序列输入之外,ESMFold2 也支持结合 MSA(Multiple Sequence Alignment,多序列比对) 进行结构预测。MSA 能够为模型提供进化背景信息,帮助模型更好地理解蛋白质序列在不同物种和同源蛋白中的演化规律,从而显著提升结构预测的准确性。
MSA 的核心价值在于,它可以揭示不同同源序列之间的保守性和共进化关系。某些残基如果在进化过程中高度保守,往往提示它们可能对蛋白质结构稳定性或功能具有重要意义;而一些残基如果在不同序列中呈现协同变化,则可能暗示它们在空间结构中相互接近或存在功能关联。通过利用这些进化约束信息,模型可以更准确地识别结构关键残基、判断潜在空间接触,并改善对不确定区域或复杂构象的预测效果。
在本示例中,我们以 泛素(ubiquitin) 作为演示对象。泛素是一种由 76 个氨基酸组成的小型调控蛋白,广泛存在于几乎所有真核生物中,在蛋白质降解、细胞信号转导、DNA 修复等过程中发挥重要作用。这里使用的 MSA 来自 ColabFold 的 MMseqs2 搜索结果,属于真实的多序列比对数据。一般来说,MSA 对具有大量同源序列的蛋白质尤其有帮助,可以用于识别保守结构特征、提高不确定区域的预测精度,并在存在多种可能构象时帮助模型解析更合理的折叠状态。
下载msa:
# download the MSA
!wget -q "https://drive.google.com/uc?export=download&id=1la9UUK_FnFQFR9VB35DIB797d87Zwm0b" -O ubiquitin.a3m
预测:
from esm.utils.msa import MSA
from esm.utils.structure.input_builder import ProteinInput, StructurePredictionInput
ubiquitin_sequence = (
"MQIFVKTLTGKTITLEVEPSDTIENVKAKIQDKEGIPPDQQRLIFAGKQLEDGRTLSDYNIQKESTLHLVLRLRGG"
)
msa = MSA.from_a3m(path="ubiquitin.a3m", remove_insertions=True, max_sequences=1000)
print(f"MSA depth: {msa.depth} sequences, length: {msa.seqlen} residues")
# Sanity check — first MSA seq should match the query
print("First MSA seq matches query:", msa.sequences[0] == ubiquitin_sequence)
print("First MSA seq:", repr(msa.sequences[0][:80]))
print("Query: ", repr(ubiquitin_sequence))
fold_result = client.fold_all_atom(
StructurePredictionInput(
sequences=[ProteinInput(id="A", sequence=ubiquitin_sequence, msa=msa)]
)
)
print(f"\npTM: {fold_result.ptm:.3f}")
print(f"Average pLDDT: {fold_result.plddt.mean().item():.1f}")
with open("ubiquitin_with_msa.cif", "w") as f:
f.write(fold_result.complex.to_mmcif())
输出:

可视化结果:
07总结
所以这么测试下来,ESMFold2 的主要优点在于它具有较强的通用结构预测能力,不仅可以预测普通蛋白质结构,还能够处理蛋白质-核酸复合物、蛋白质-小分子体系、非标准氨基酸、共价修饰以及结合 MSA 信息的复杂建模任务;同时,它可以进行全原子级别的结构预测,相比只输出蛋白质主链或单链结构的方法,更适合用于真实生物分子复合物、治疗性多肽和修饰分子体系的建模研究。此外,ESMFold2 支持单序列快速预测,也可以通过增加折叠迭代次数、扩散采样步数或引入 MSA 来提高预测精度,具有较好的灵活性。
关于缺点,再慢慢测试吧,哈哈哈。谢谢各位大佬们的点赞和分享~
一些reference:
https://biohub.org/news/world-model-of-protein-biology/
https://huggingface.co/biohub/ESMFold2
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-31,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号