Dnaapler实战:如何统一环状微生物基因组的起始位置

Dnaapler实战:如何统一环状微生物基因组的起始位置

用户1075469
发布于 2026-06-24 12:12:23
发布于 2026-06-24 12:12:23
Hello,Hello小伙伴们大家好,我是小编豆豆。近年来,随着三代测序和长读长组装技术的发展,越来越多完整的环状基因组被成功组装出来。除了常见的细菌染色体和质粒,噬菌体、古菌等环状基因组也逐渐成为微生物组和病毒组研究中的重要对象。然而,环状基因组虽然首尾相连,但在实际分析中仍然需要一个相对统一的起始位置。若不同基因组的起点不一致,可能会影响后续注释结果整理、基因组比较、共线性展示以及批量分析的规范性。今天给大家介绍一款可以用于细菌、质粒、噬菌体和古菌环状基因组起点校正的自动化工具——Dnaapler。
工作流程
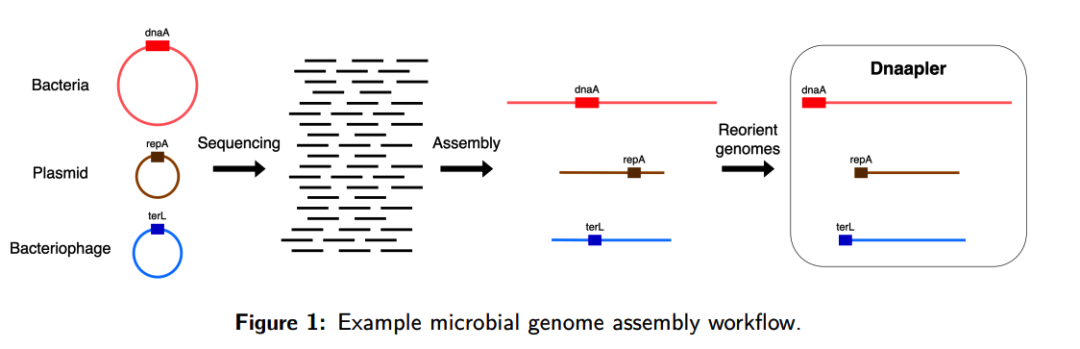
流程解读
(A)输入为已经组装完成的环状核苷酸序列,支持 FASTA 或 GFA 格式;若输入为 GFA 文件,Dnaapler 只处理符合环状结构特征的序列,其余序列保持不变;
(B)根据序列类型选择对应的重定向模式,例如细菌染色体可使用 dnaapler chromosome,质粒可使用 dnaapler plasmid,噬菌体可使用 dnaapler phage,混合序列可使用 dnaapler all;
(C)Dnaapler 通过 blastx 将输入核苷酸序列与氨基酸序列数据库进行比对,寻找目标起始标记基因,例如细菌染色体中的 dnaA、质粒中的 repA 以及噬菌体中的 terL;
(D)在识别到候选起始基因后,软件会检查该基因的起始密码子是否存在,并在重定向后使该基因位于正向链上;
(E)若目标起始基因满足要求,Dnaapler 会将环状基因组旋转到该基因位置开始输出,从而统一环状基因组的起始位置,减少随机断点对基因注释、移动遗传元件识别、泛基因组分析和基因组可视化的影响。
Github:
https://github.com/gbouras13/dnaapler
官方文档:
https://dnaapler.readthedocs.io/en/latest/
程序子命令:
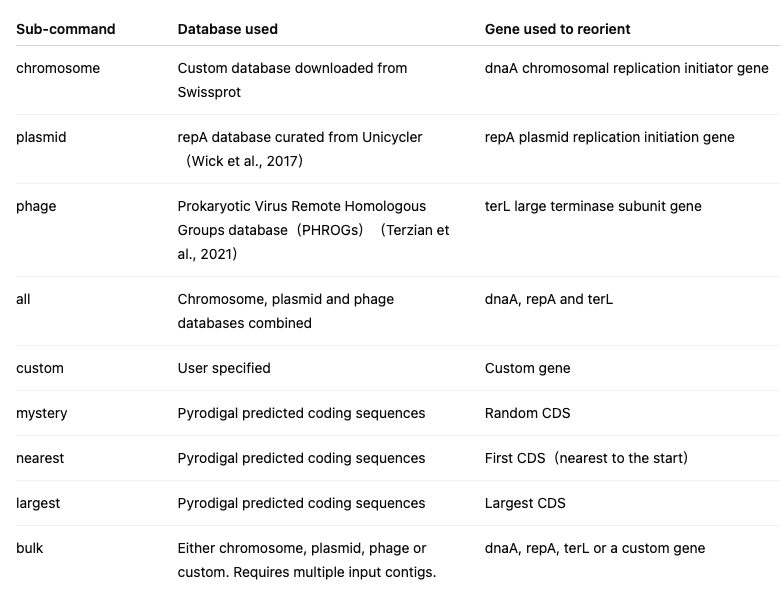
说明:
Dnaapler 的 chromosome、phage 和 plasmid 子命令使用 blastx 进行搜索,即利用翻译后的核苷酸序列作为查询序列,在蛋白数据库中分别检索 dnaA、terL 或 repA 基因。all 子命令会同时针对上述三个数据库运行 blastx 搜索,并在输入基因组中同时命中 dnaA 和 repA 的情况下,优先选择 dnaA。
软件安装
# 创建一个名为Dnaapler的conda环境,并安装dnaapler
conda create -n Dnaapler -c conda-forge -c bioconda dnaapler
# conda install 直接安装
conda install -c bioconda dnaapler
# pip install 安装
pip install dnaapler
# 激活环境
conda activate Dnaapler 安装说明:
小编的安装更推荐第一种方法,因为小编习惯将每个软件单独创建一个虚拟conda环境,避免软件依赖产生冲突。
使用方法
dnaapler -h
Usage: dnaapler [OPTIONS] COMMAND [ARGS]...
Options:
-h, --help Show this message and exit.
-V, --version Show the version and exit.
Commands:
all Reorients contigs to begin with any of dnaA, repA, terL or
archaeal COG1474 Orc1/cdc6
archaea Reorients your genome to begin with the archaeal COG1474
Orc1/cdc6 origin recognition complex gene
bulk Reorients multiple genomes to begin with the same gene
chromosome Reorients your genome to begin with the dnaA chromosomal
replication initiation gene
citation Print the citation(s) for this tool
custom Reorients your genome with a custom database
largest Reorients your genome the begin with the largest CDS as called
by pyrodigal
mystery Reorients your genome with a random CDS
nearest Reorients your genome the begin with the first CDS as called by
pyrodigal
phage Reorients your genome to begin with the terL large terminase
subunit
plasmid Reorients your genome to begin with the repA replication
initiation gene主要参数解读
自动识别并重定向多种类型的 contig,可识别 dnaA、repA、terL 或古菌 COG1474 Orc1/cdc6dnaapler all -h
Usage: dnaapler all [OPTIONS]
Options:
-h, --help Show this message and exit.
-V, --version Show the version and exit.
-i, --input PATH Path to input file in FASTA or GFA format
[required]
-o, --output PATH Output directory [default: output.dnaapler]
-t, --threads INTEGER Number of threads to use with MMseqs2 [default: 1]
-p, --prefix TEXT Prefix for output files [default: dnaapler]
-f, --force Force overwrites the output directory
-e, --evalue TEXT E-value for MMseqs2 [default: 1e-10]
--ignore TEXT Text file listing contigs (one per row) that are to
be ignored OR comma separated list of contig names
to ignore OR '-' to read from stdin
--db, --database TEXT Lets you choose a subset of databases rather than
all 4. Must be one of: 'all', 'dnaa', 'repa',
terl', 'cog1474', 'dnaa,repa', 'dnaa,terl',
'repa,terl', 'dnaA,cog1474', 'cog1474,terl',
'cog1474,repa', 'dnaa,cog1474,repa',
'dnaa,cog1474,terl' or 'cog1474,repa,terl'
[default: all]
-c, --custom_db PATH FASTA file with amino acids that will be used as a
custom MMseqs2 database to reorient your sequence
however you want.
-a, --autocomplete TEXT Choose an option to autocomplete reorientation if
MMseqs2 based approach fails. Must be one of: none,
mystery, largest, or nearest [default: none]
--seed_value INTEGER Random seed to ensure reproducibility. [default:
13]查看 dnaapler all 的帮助信息说明:其他子命名的参数大同小异,读者可以借助AI协助自己理解含义,这里不再赘述
实战演练
# 统一细菌环状染色体的起始位置,以 dnaA 基因为起点进行重定向
dnaapler chromosome -i chromosome.fasta -o C333 -t 8 -p C333 -f
# 自动识别输入序列中的细菌染色体、质粒、噬菌体或古菌环状 contig,并统一其起始位置
dnaapler all -i scoffold.fasta -o C333 -t 8 -p C333 -f
# 将噬菌体环状基因组重定向至 terL 大末端酶亚基基因位置
dnaapler phage -i sa3int_phage.fasta -o sa3int -t 8 -p sa3int -f核心结果解读
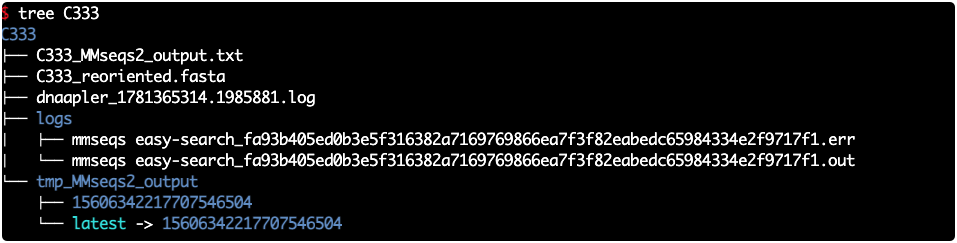
最重要的结果文件,这是重定向后的 FASTA 序列说明:Dnaapler 子命令较多,本文仅介绍其中较常用的核心命令,起到抛砖引玉的作用。其他命令和参数,读者可结合官方文档、AI 辅助及实际项目需求灵活使用。
参考文献
- George B, Grigson S R, Papudeshi B, et al. Dnaapler: A tool to reorient circular microbial genomes[J]. Journal of Open Source Software, 2024, 9(93): 5968. DOI: 10.21105/joss.05968.
- Altschul S F, Gish W, Miller W, et al. Basic local alignment search tool[J]. Journal of Molecular Biology, 1990, 215(3): 403-410. PMID: 2231712. DOI: 10.1016/S0022-2836(05)80360-2.
- Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets[J]. Nature Biotechnology, 2017, 35(11): 1026-1028. DOI: 10.1038/nbt.3988.
- Larralde M. Pyrodigal: Python bindings and interface to Prodigal, an efficient method for gene prediction in prokaryotes[J]. Journal of Open Source Software, 2022, 7(72): 4296. DOI: 10.21105/joss.04296.
- Hyatt D, Chen G L, LoCascio P F, et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification[J]. BMC Bioinformatics, 2010, 11: 119. DOI: 10.1186/1471-2105-11-119.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-14,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号