课前准备--空间组学解析肿瘤异质性
原创
课前准备--空间组学解析肿瘤异质性
原创
追风少年i
发布于 2026-06-22 08:20:53
发布于 2026-06-22 08:20:53
作者,Evil Genius
2026年空间转录组系列课程已经在进行中,各位学员上完课程后,课程上提到的文献一定要亲自看一看,提供给大家的脚本亲自做一做,遇到问题群里随时答疑。
今天我们来汇总一些空间平台解析肿瘤内部异质性。
核心问题:肿瘤内异质性 (ITH)
定义:单个肿瘤内存在多种多样的癌细胞状态、基因型和微环境生态位。
重要性:是治疗耐药和疾病进展的主要驱动因素。
本质:肿瘤是长期进化、突变累积和克隆选择的结果;是一个包含癌细胞、免疫细胞和基质细胞的动态、可塑的生态系统。
技术演进
古典病理学:首次发现组织学异质性。
DNA测序:提供分子层面的ITH见解,揭示克隆性肿瘤发生。
单细胞基因组学:以前所未有的粒度识别新细胞亚型和状态。
局限性: 这些方法需要组织解离,丢失了关键的空间位置信息。
解决方案:空间生物学的出现,因为细胞行为受空间组织控制。
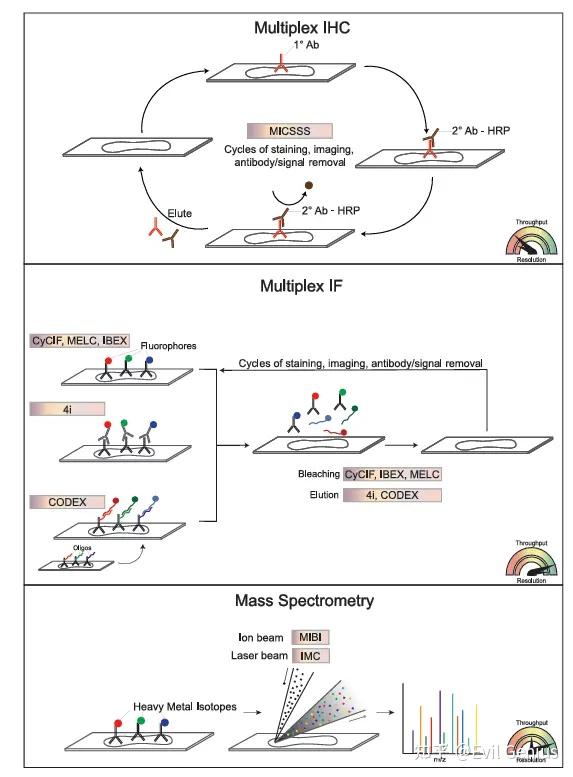
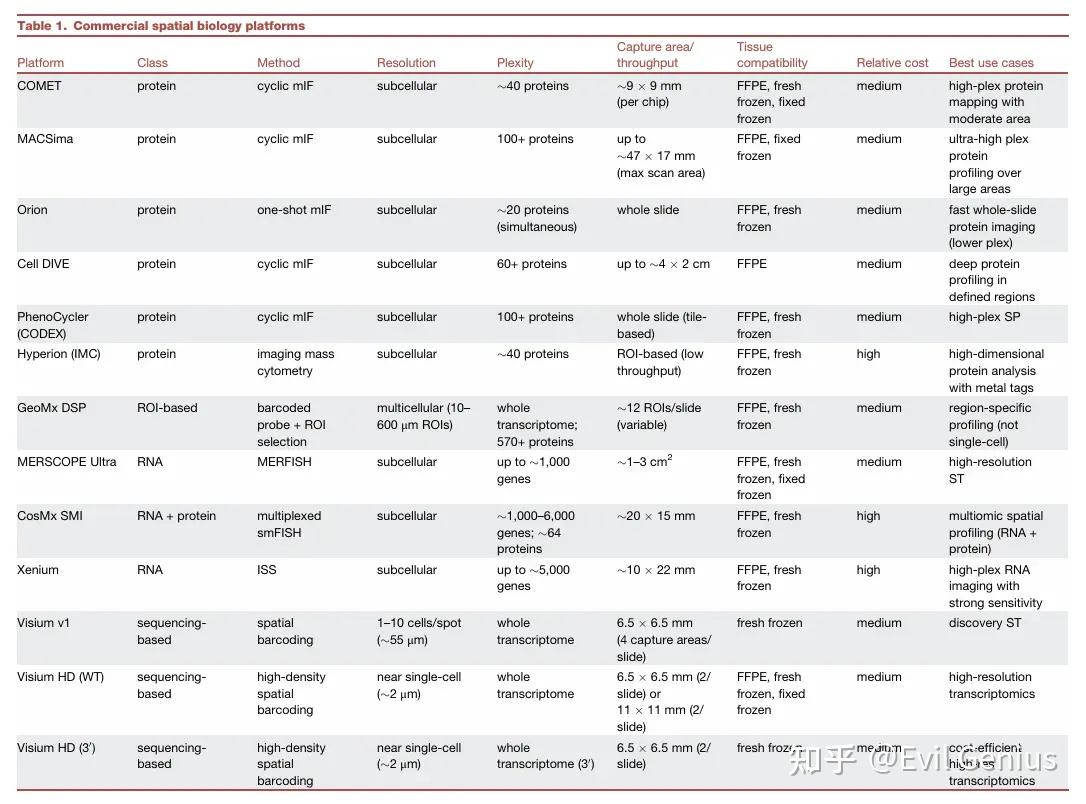
空间蛋白质组学 (SP)
原理: 基于抗体检测(类似IHC/IF),但具备高度多重检测能力(可同时检测50-100个标记物)。
三大类别:
多重IHC (mIHC):
方法:连续染色、成像、抗体剥离/失活循环(如MICSSS)。
优势:使用标准明场显微镜,兼容FFPE组织。
局限:循环次数有限(~20轮)。
多重IF (mIF):
方法:
循环法:迭代染色、成像、失活(如CyCIF, t-CyCIF, IBEX, seqIF, MACSima)。
一次性法:使用DNA条形码抗体,通过多轮杂交和成像解码(如CODEX)。
优势:单细胞分辨率,更好的多重能力(40-100+蛋白)。
仪器示例:CODEX, Cell DIVE, PhenoCycler, Orion, MACSima, COMET。
基于质谱 (MS):
方法:使用金属同位素标签,通过质谱检测(如IMC)或离子束成像(如MIBI)。
优势:无光谱重叠,背景低,高分辨率。
局限:仪器昂贵且需专业知识,扫描面积/速度有限,通常需要冷冻或固定冷冻组织(IMC/MIBI I)。
选择依据: 取决于实验需求、组织类型(FFPE vs 冷冻)、预算、通量和分辨率要求。
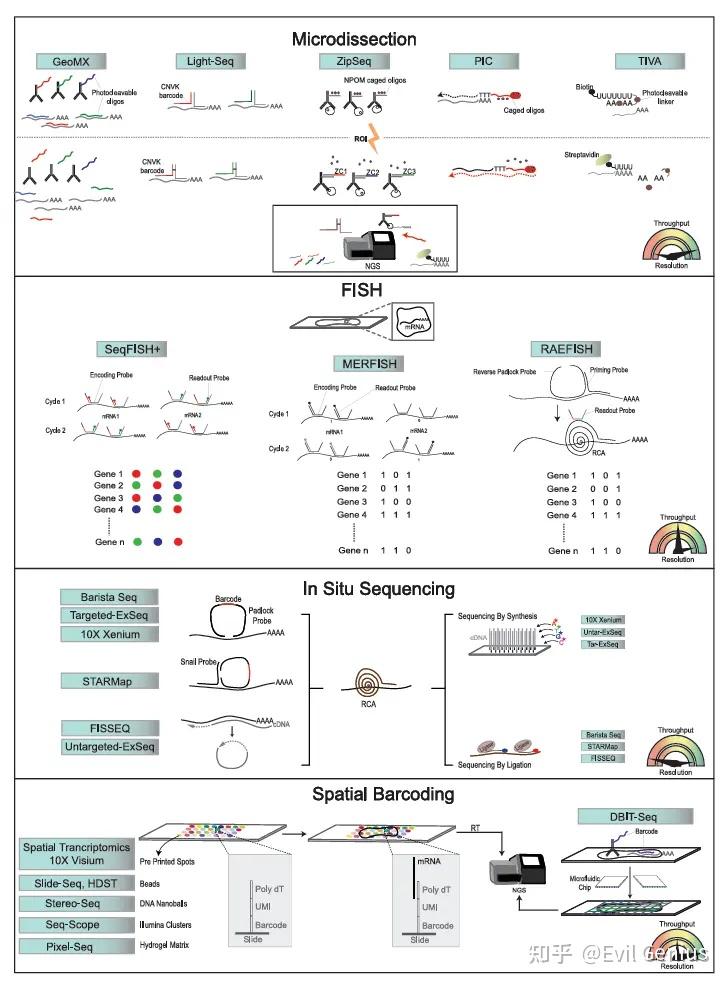
空间转录组学 (ST)
核心挑战: 在检测RNA的同时保留其空间位置信息(RNA测序通常需要组织裂解)。
四大类别:
显微切割/ROI-based (基于感兴趣区域):
方法:使用激光或光控技术分离特定区域RNA(如GeoMx DSP, Light-seq, ZipSeq, TIVA, PIC)。
特点:非单细胞分辨率,提供选定区域内的全转录组或靶向数据。
基于荧光原位杂交 (FISH):
方法:使用多轮荧光探针杂交、成像和剥离来解码RNA(如seqFISH, MERFISH)。
商业平台:Vizgen MERSCOPE (MERFISH), Nanostring CosMx (smFISH)。
特点:真正的单细胞/亚细胞分辨率,灵敏度高,但基因数量受光学拥挤限制(目前~1000-6000个基因)。需要预选基因组合。
新方法:RAEFISH 通过RCA扩增,实现全转录组覆盖,但灵敏度可能有折衷;PRISM 利用强度区分条形码,降低设备要求。
基于原位测序 (ISS):
方法:在组织原位对RNA进行测序(如STARmap, BaristaSeq, ExSeq, FISSEQ, Xenium)。
特点:直接读取序列信息,单细胞/亚细胞分辨率,但同样受光学拥挤影响,基因通量有限。
基于空间条形码 (Spatial Barcoding):
方法:使用带有位置条形码的探针阵列捕获mRNA,通过测序将转录本映射回空间位置(如Visium, Slide-seq, HDST, Stereo-seq, DBiT-seq)。
商业平台:10x Genomics Visium。
特点:全转录组覆盖,流程与现有测序兼容,无需专用成像设备。但非单细胞分辨率(每个点可能包含多个细胞),最新版本(Visium HD)接近单细胞。
空间功能基因组学
目的: 将基因型(扰动)与表型(空间位置)直接关联,实现反向遗传学(从基因到功能)。
方法: 结合CRISPR筛选与空间读数。
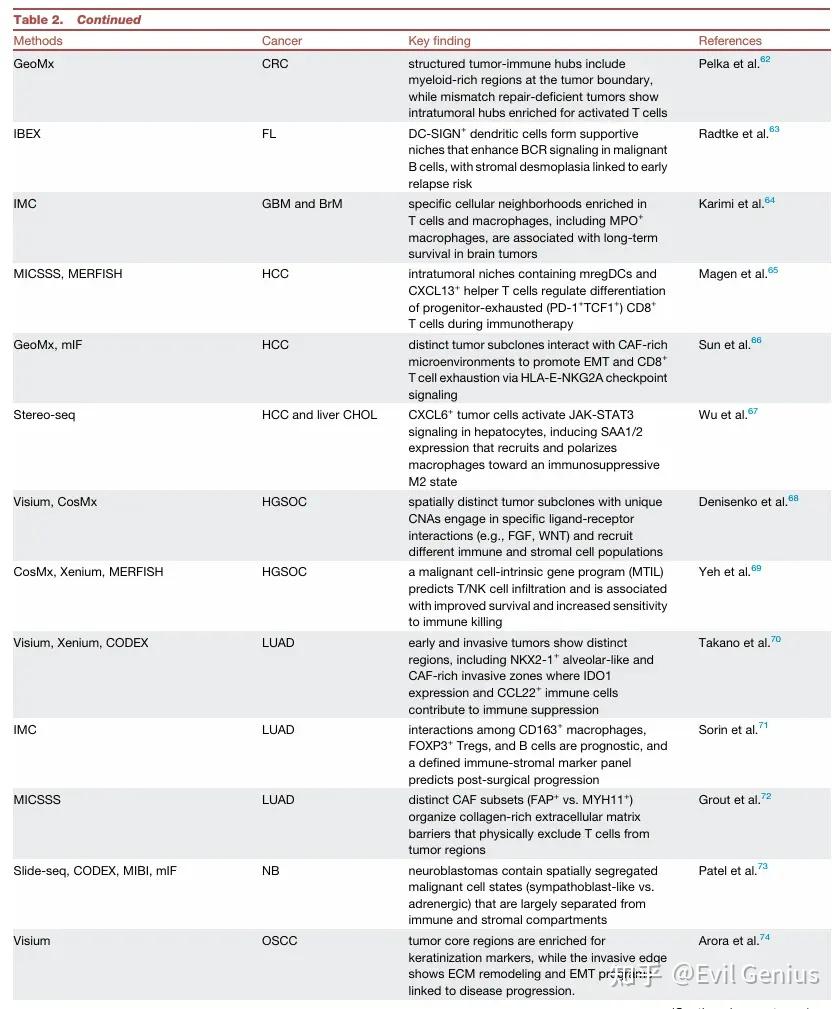
基于蛋白质读数 (SP-readout):
使用蛋白条形码(如Pro-Codes, EpicTag, EPICode)标记携带特定sgRNA的细胞。
通过抗体染色检测条形码和表型标记物。
关键方法:Perturb-map。应用:发现TGFBR2缺失、IL-4、CCL7、PAI1/2等基因在肿瘤微环境重塑和免疫逃逸中的作用。
基于转录组读数 (ST-readout):
在ST平台上直接检测sgRNA或扰动引起的转录组变化。
关键方法: Perturb-FISH (MERFISH), Perturb-Multi (CROPseq-multi + MERFISH/ISS), CRISPRmap (seqFISH), PerturbView (ISS), Perturb-CAST (Visium), Perturb-DBiT (DBiT-seq)。
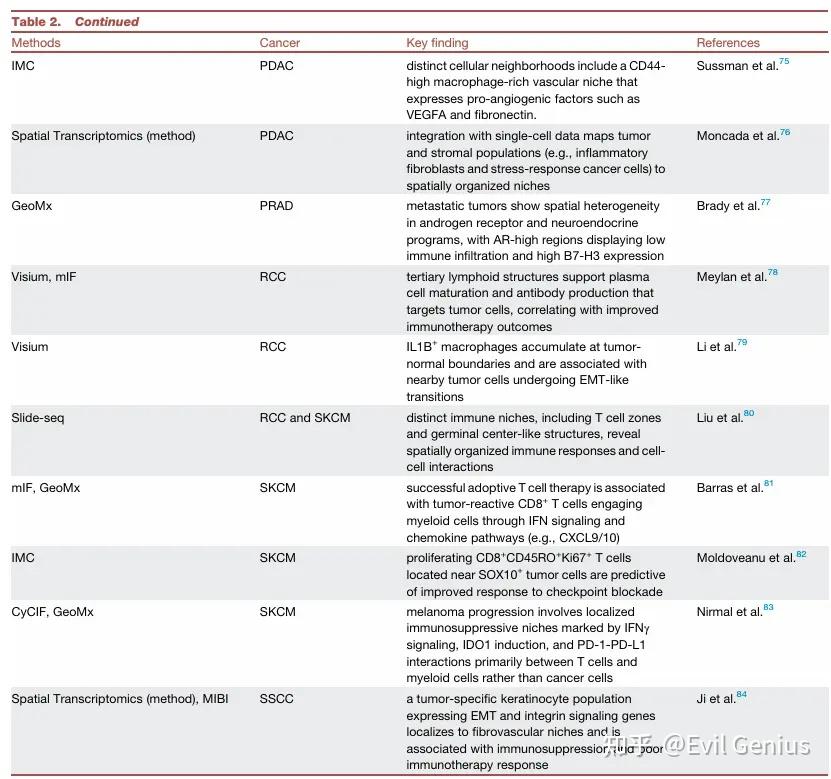
空间克隆追踪
目的: 重建肿瘤内不同克隆谱系的进化历史和空间分布。
方法类别:
利用内源性体细胞突变:
方法:显微切割+测序,Slide-DNA-seq,Slide-tags。
优势:适用于人类临床样本(无需工程改造)。
特点:Slide-tags可实现单细胞核分辨率的空间转录组/表观基因组分析。
利用工程化条形码:
整合酶系统 (intMEMOIR): 通过DNA重组记录谱系,可结合FISH。
慢病毒条形码 (TREX, SpaceBar): 随机序列标记细胞,可结合ST。
利用CRISPR诱导的“疤痕”:
原理:诱导Cas9在条形码上产生随机突变(插入/缺失),形成独特标识。
方法:scGESTALT, ScarTrace, LINNAEUS, macsGESTALT, iTracer, Zombie。
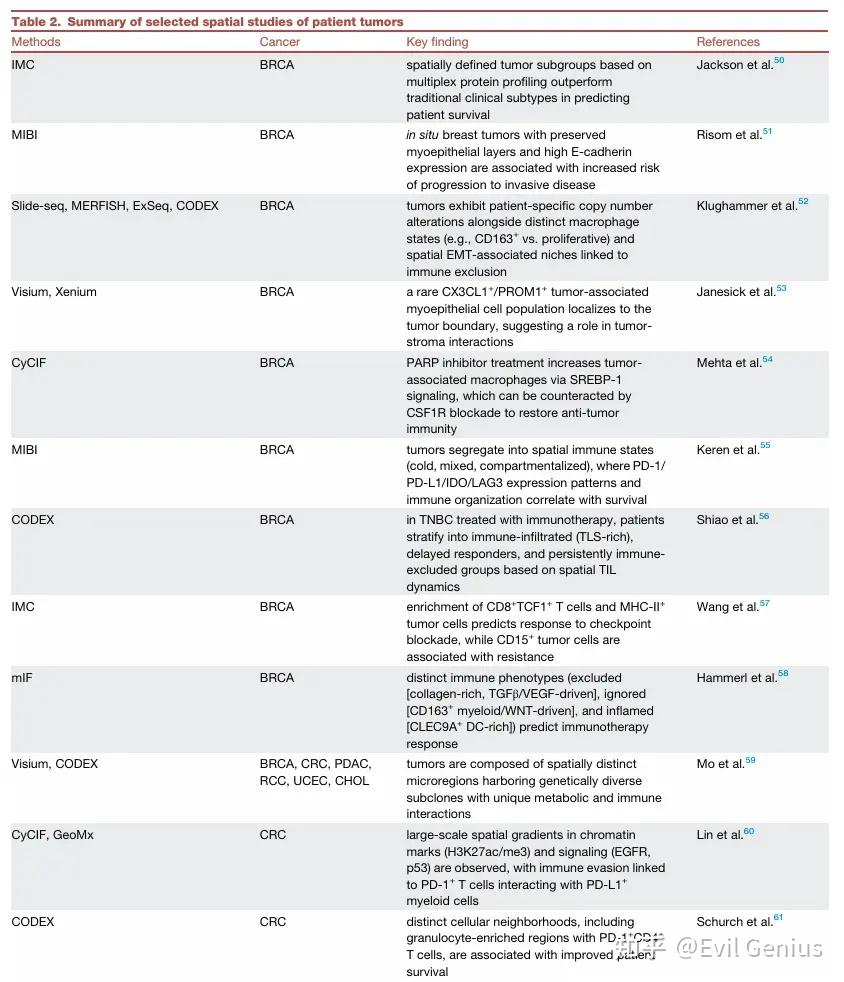
关键应用 (macsGESTALT): 在胰腺癌中发现,大多数克隆不转移,而获得杂交EMT状态和表达S100基因的罕见克隆具有转移能力。
综合模型: 如KP-Tracer(结合CRISPR疤痕和ST),用于研究肺癌进展中的空间群落和克隆动态。
整合多组学与计算挑战
多模态整合: 结合SP、ST、功能基因组学、克隆追踪以及表观基因组学、代谢组学是未来方向。
新兴技术: spatial-CITE-seq (RNA+蛋白), SPOTS (RNA+蛋白), 空间表观-转录组共测序等。
计算挑战:
数据稀疏性、高噪声、平台间批次效应。
细胞分割(识别细胞边界)困难。
数据整合和空间统计分析复杂。
解决方案:
开发和使用开放、可扩展的分析框架:SquidPy, SCIMAP, Giotto, SpatialData, Seurat。
人工智能(AI)的应用:加速模式识别、数据插补、从H&E染色预测ST数据。
临床转化与未来展望
当前临床生物标志物局限: 常规IHC(如PD-L1)预测免疫治疗反应的能力有限。
空间生物标志物的潜力:
巨噬细胞PD-L1表达比肿瘤细胞PD-L1更能预测ICI反应。
特定的免疫细胞三合会(如CXCL13+ Th, PD-1hi 祖CD8 T, 成熟DCs)与治疗响应相关。
特定的细胞空间邻近性与患者生存相关。
特定巨噬细胞状态(IL4I1+ 有利;SPP1+ 不利)和三级淋巴结构(TLS)是重要的预后/预测指标。
临床转化的挑战:
成本:仪器和试剂昂贵。
基础设施:需要大规模数据存储和计算能力。
专业知识:缺乏生物信息学专业人才。
技术权衡:通量与分辨率之间的矛盾。
标准化:不同平台和实验室间的方案和结果需要统一。
未来方向:
技术将更便宜、更易用、分辨率更高。
与AI的深度融合,将加速基础研究和临床诊断。
有望在未来临床实践中,通过AI分析H&E染色结合低多重空间蛋白检测来指导治疗。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号