分析梳理--分子动力学模拟的常规步骤四(Gromacs)
原创
分析梳理--分子动力学模拟的常规步骤四(Gromacs)
原创
追风少年i
发布于 2026-04-21 14:59:09
发布于 2026-04-21 14:59:09
作者,Evil Genius
今天我们继续分子动力学:能量最小化
也是需要分两步进行
首先我们要学习进行MD的参数设置,em_real.mdp
;em_real.mdp-grompp的参数输入文件
integrator = steep;指定使用最陡下降法进行能量最小化.若设为cg则使用共轭梯度法;
emtol = 1000.0 ;若力的最大值小于此值则认为能量最小化收敛k/mol/nm.
emstep=0.01;初始步长
nsteps= 50000;在能量最小化中,指定最大迭代次数
; 近邻列表, 相互作用计算参数
nstlist = 1 ; 更新近邻列表的频率. 1 表示每步都更新
cutoff-scheme = Verlet ; 生成带有缓冲的对列表。 (5.1后) 没有水更快
ns_type = grid ; 近邻列表的确定方法,适用于大体系
coulombtype = PME ; 计算长程静电的方法.PME为粒子网格 Ewald 方法
rcoulomb = 1.0 ; 长程库仑力的截断值
rvdw = 1.0 ; 范德华距离截断值
pbc = xyz ; 在所有方向上使用周期性边界条件。其中;后面是注释信息。
首先能量最小化的第一步:生成能量最小化的tpr文件
gmx grompp -f em_real.mdp -c solv_ions.gro -p topol.top -o em.tpr第二步:执行能量最小化
gmx mdrun
看看所有参数
Options to specify input files:
-s [<.tpr>] (topol.tpr)
Portable xdr run input file
-cpi [<.cpt>] (state.cpt) (Opt.)
Checkpoint file
-table [<.xvg>] (table.xvg) (Opt.)
xvgr/xmgr file
-tablep [<.xvg>] (tablep.xvg) (Opt.)
xvgr/xmgr file
-tableb [<.xvg> [...]] (table.xvg) (Opt.)
xvgr/xmgr file
-rerun [<.xtc/.trr/...>] (rerun.xtc) (Opt.)
Trajectory: xtc trr cpt gro g96 pdb tng
-ei [<.edi>] (sam.edi) (Opt.)
ED sampling input
-multidir [<dir> [...]] (rundir) (Opt.)
Run directory
-awh [<.xvg>] (awhinit.xvg) (Opt.)
xvgr/xmgr file
-plumed [<.dat>] (plumed.dat) (Opt.)
Generic data file
-membed [<.dat>] (membed.dat) (Opt.)
Generic data file
-mp [<.top>] (membed.top) (Opt.)
Topology file
-mn [<.ndx>] (membed.ndx) (Opt.)
Index file
Options to specify output files:
-o [<.trr/.cpt/...>] (traj.trr)
Full precision trajectory: trr cpt tng
-x [<.xtc/.tng>] (traj_comp.xtc) (Opt.)
Compressed trajectory (tng format or portable xdr format)
-cpo [<.cpt>] (state.cpt) (Opt.)
Checkpoint file
-c [<.gro/.g96/...>] (confout.gro)
Structure file: gro g96 pdb brk ent esp
-e [<.edr>] (ener.edr)
Energy file
-g [<.log>] (md.log)
Log file
-dhdl [<.xvg>] (dhdl.xvg) (Opt.)
xvgr/xmgr file
-field [<.xvg>] (field.xvg) (Opt.)
xvgr/xmgr file
-tpi [<.xvg>] (tpi.xvg) (Opt.)
xvgr/xmgr file
-tpid [<.xvg>] (tpidist.xvg) (Opt.)
xvgr/xmgr file
-eo [<.xvg>] (edsam.xvg) (Opt.)
xvgr/xmgr file
-px [<.xvg>] (pullx.xvg) (Opt.)
xvgr/xmgr file
-pf [<.xvg>] (pullf.xvg) (Opt.)
xvgr/xmgr file
-ro [<.xvg>] (rotation.xvg) (Opt.)
xvgr/xmgr file
-ra [<.log>] (rotangles.log) (Opt.)
Log file
-rs [<.log>] (rotslabs.log) (Opt.)
Log file
-rt [<.log>] (rottorque.log) (Opt.)
Log file
-mtx [<.mtx>] (nm.mtx) (Opt.)
Hessian matrix
-if [<.xvg>] (imdforces.xvg) (Opt.)
xvgr/xmgr file
-swap [<.xvg>] (swapions.xvg) (Opt.)
xvgr/xmgr file
Other options:
-deffnm <string>
Set the default filename for all file options
-xvg <enum> (xmgrace)
xvg plot formatting: xmgrace, xmgr, none
-dd <vector> (0 0 0)
Domain decomposition grid, 0 is optimize
-ddorder <enum> (interleave)
DD rank order: interleave, pp_pme, cartesian
-npme <int> (-1)
Number of separate ranks to be used for PME, -1 is guess
-nt <int> (0)
Total number of threads to start (0 is guess)
-ntmpi <int> (0)
Number of thread-MPI ranks to start (0 is guess)
-ntomp <int> (0)
Number of OpenMP threads per MPI rank to start (0 is guess)
-ntomp_pme <int> (0)
Number of OpenMP threads per MPI rank to start (0 is -ntomp)
-pin <enum> (auto)
Whether mdrun should try to set thread affinities: auto, on, off
-pinoffset <int> (0)
The lowest logical core number to which mdrun should pin the first
thread
-pinstride <int> (0)
Pinning distance in logical cores for threads, use 0 to minimize
the number of threads per physical core
-gpu_id <string>
List of unique GPU device IDs available to use
-gputasks <string>
List of GPU device IDs, mapping each task on a node to a device.
Tasks include PP and PME (if present).
-[no]ddcheck (yes)
Check for all bonded interactions with DD
-rdd <real> (0)
The maximum distance for bonded interactions with DD (nm), 0 is
determine from initial coordinates
-rcon <real> (0)
Maximum distance for P-LINCS (nm), 0 is estimate
-dlb <enum> (auto)
Dynamic load balancing (with DD): auto, no, yes
-dds <real> (0.8)
Fraction in (0,1) by whose reciprocal the initial DD cell size will
be increased in order to provide a margin in which dynamic load
balancing can act while preserving the minimum cell size.
-nb <enum> (auto)
Calculate non-bonded interactions on: auto, cpu, gpu
-nstlist <int> (0)
Set nstlist when using a Verlet buffer tolerance (0 is guess)
-[no]tunepme (yes)
Optimize PME load between PP/PME ranks or GPU/CPU
-pme <enum> (auto)
Perform PME calculations on: auto, cpu, gpu
-pmefft <enum> (auto)
Perform PME FFT calculations on: auto, cpu, gpu
-bonded <enum> (auto)
Perform bonded calculations on: auto, cpu, gpu
-update <enum> (auto)
Perform update and constraints on: auto, cpu, gpu
-[no]v (no)
Be loud and noisy
-pforce <real> (-1)
Print all forces larger than this (kJ/mol nm)
-[no]reprod (no)
Avoid optimizations that affect binary reproducibility; this can
significantly reduce performance
-cpt <real> (15)
Checkpoint interval (minutes)
-[no]cpnum (no)
Keep and number checkpoint files
-[no]append (yes)
Append to previous output files when continuing from checkpoint
instead of adding the simulation part number to all file names
-nsteps <int> (-2)
Run this number of steps (-1 means infinite, -2 means use mdp
option, smaller is invalid)
-maxh <real> (-1)
Terminate after 0.99 times this time (hours)
-replex <int> (0)
Attempt replica exchange periodically with this period (steps)
-nex <int> (0)
Number of random exchanges to carry out each exchange interval (N^3
is one suggestion). -nex zero or not specified gives neighbor
replica exchange.
-reseed <int> (-1)
Seed for replica exchange, -1 is generate a seedgmx mdrun -v -deffnm em
-V可以看到运行结果:它使mdrun输出更多信息,这样就会在屏幕上输出每步运行的情况。
-deffnm<string>为所有文件选项设置默认文件名,选项定义了输入文件和输出文件的名称。
我们将得到以下文件:
em.log:ASCll文本的日志文件,记录了能量最小化过程
em.edr:二进制能量文件
em.trr:全精度的二进制轨迹文件
em.gro:能量最小化后的结构
我们来运行一下
gmx mdrun -ntmpi 4 -ntomp 17 -v -deffnm em能量最小化是否成功有两个重要的指标:第一个是势能(在能量最小化过程屏幕上的最后输出)。Epot应当是负值,根据体系大小和水分子的多少,大约在105-106的数量级(对水中的单个蛋白质而言)。
第二个重要的指标是力的最大值Fmax.我们在em_real.mdp中设置的最大值是emtol=1000.0,这表示Fmax的目标值不能大于1000kJ * mol-1nm-1。能量最小化完成后,你有可能得到一个合理的Epot,但Fmax>emtol.。如果是这样,用于模拟的体系可能不够稳定,模拟过程中体系会崩溃。可能需要更改一下能量最小化的参数设置如,方法、最大步数等,需要重新运行grompp。
最后看一下生成的结构
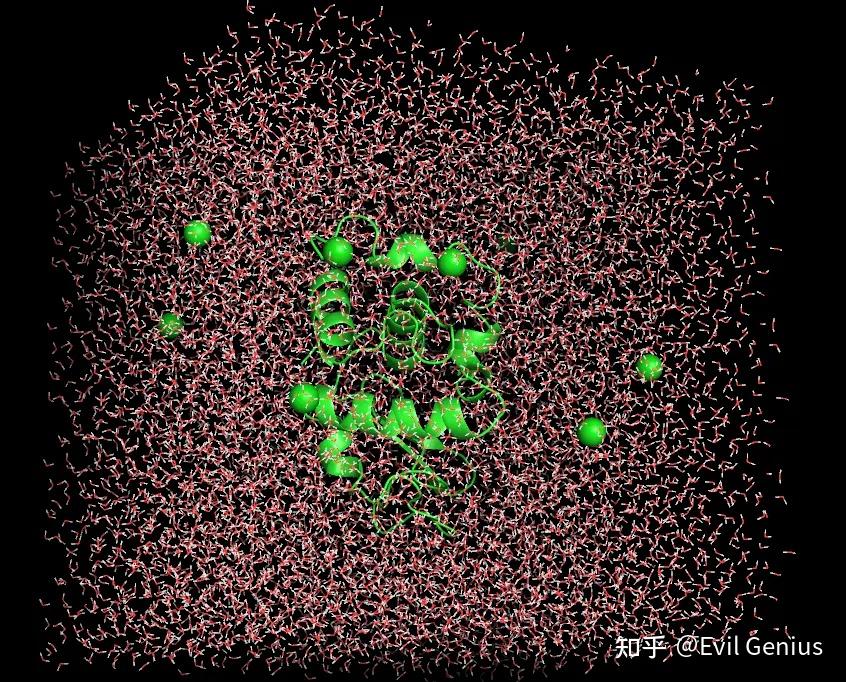
绘图看看能量变化
gmx energy -f em.edr -o potential.xvg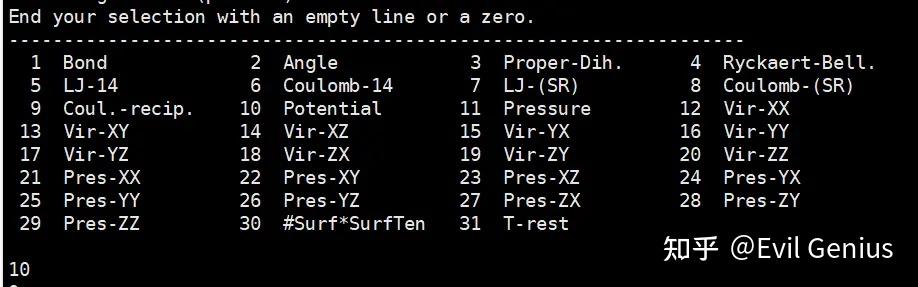
其中这些内容的意义
能量与相互作用项
这些是构成系统总势能的不同贡献部分。
编号 | 名称 | 物理含义 | 备注 |
|---|---|---|---|
1 | Bond | 键长伸缩能 | 原子间化学键被拉伸或压缩产生的能量。 |
2 | Angle | 键角弯折能 三个原子形成的化学键角度变化产生的能量。 | |
3 | Proper-Dih. | 正常二面角能 | 沿化学键旋转四个原子时,由特定二面角产生的能量。 |
4 | Ryckaert-Bell. | RB二面角能 | 一种特殊的二面角函数形式,常用于烷烃链。 |
5 | LJ-14 | 1-4相互作用LJ能 | 相隔三个化学键的原子对之间,范德华作用能量。 |
6 | Coulomb-14 | 1-4相互作用静电能 | 相隔三个化学键的原子对之间,静电作用能量。 |
7 | LJ-(SR) | 短程LJ能 | 在截断距离内的所有非键原子对的范德华能量总和。 |
8 | Coulomb-(SR) | 短程静电能 | 在截断距离内的所有非键原子对的静电能量总和。 |
9 | Coul.-recip. | 倒易空间静电能 | 使用PME方法计算的长程静电贡献部分。 |
总能量与宏观性质项
编号 | 名称 | 物理含义 | 备注 |
|---|---|---|---|
10 | Potential | 系统总势能 | 这是最重要的项。等于上述1-9项的总和 (Bond + Angle + ... + Coul.-recip.)。 |
11 | Pressure | 系统总压力 | 由动能和位力计算得到,是标量压力(各向同性时)。 |
12-20 | Vir-XX ... Vir-ZZ | 位力张量分量 | 用于计算压力的张量,对角线(XX, YY, ZZ)和非对角线(XY等)。 |
21-29 | Pres-XX ... Pres-ZZ | 压力张量分量 | 也是压力张量的分量,在位力的基础上进一步计算得到。 |
30 | #Surf*SurfTen | 表面张力 | 如果有界面(如脂双层),该项表示表面张力。 |
31 | T-rest | T-rest能量 | 如果使用了温度退火或约束,该项记录相关能量。 |
在能量最小化(em)中的重点
你在做能量最小化(gmx mdrun -deffnm em),最应该关注的是:
第10项 Potential (总势能)
它应该持续下降,并最终收敛到一个稳定的负值(对于蛋白质-水体系,通常是很大的负数,例如 -1e5 到 -1e6 kJ/mol 量级)。
如果它震荡或上升,说明体系有问题(比如原子重叠严重)。
第7项 LJ-(SR) 和 第8项 Coulomb-(SR)
它们的值也应该下降并收敛。如果它们下降得非常缓慢或出现突变,可能说明范德华或静电相互作用存在严重冲突。
第11项 Pressure (压力)
在能量最小化阶段,压力值可以不那么合理(甚至很大),这很正常,因为体系还未平衡。但在NPT平衡时,它必须收敛到目标值(如1 bar)。
简单绘图看看
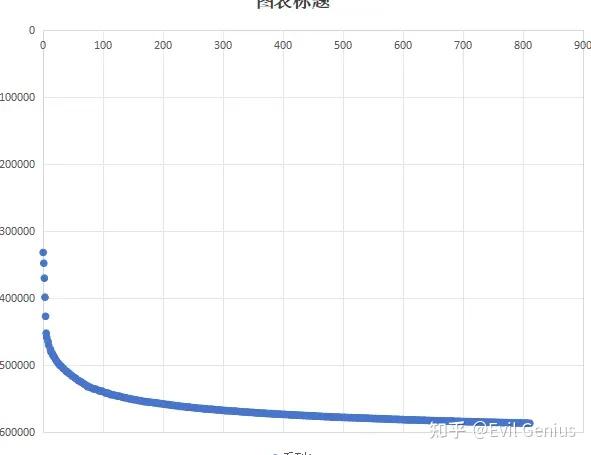
能量趋于平缓,收敛,是一个正常的能量最下化的过程。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号